
Kmenové buňky v léčbě amyotrofické laterální sklerózy – přehled současných klinických zkušeností
Stem Cell Therapy for Amyotrophic Lateral Sclerosis – an Overview of Current Clinical Experience
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease of motor neurons. While the genetics behind familial as well as sporadic ALS are being increasingly uncovered, pathophysiology remains incompletely understood and there are no effective treatment options. However, as preclinical results provided some rationale for the use of stem cells as support cells for the dying motor neurons, stem cells are being considered as a potential treatment strategy. Based on the preclinical models, translational human trials have been carried out using various types of stem cells, as well as a range of cell delivery methods. To date, no trial has demonstrated a clear therapeutic benefit. Here, we provide a critical review of current clinical trials using either mesenchymal or neural stem cells to treat ALS patients. In order to provide robust assessment of the efficacy of stem cells, it will be essential to standardize administration protocols, identify the most suitable cell type as well as to validate more reliable biomarkers of disease progression in longitudinal clinical studies.
Key words:
amyotrophic lateral sclerosis – stem cell therapy – neural progenitor cells – mesenchymal stem cells – granulocyte-colony stimulating factor – clinical trials
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
D. Baumgartner; P. Marusič; R. Mazanec
Působiště autorů:
Neurologická klinika 2. LF UK a FN Motol, Praha
Vyšlo v časopise:
Cesk Slov Neurol N 2017; 80/113(1): 27-33
Kategorie:
Přehledný referát
doi:
https://doi.org/10.14735/amcsnn201727
Souhrn
Amyotrofická laterální skleróza (ALS) je progresivní neurodegenerativní onemocnění motorických neuronů. Ačkoliv se během posledních několika let podařilo definovat genetické pozadí nejen familiárních, ale i některých sporadických forem, patofyziologie onemocnění není stále objasněna a není znám žádný účinný terapeutický postup. Na základě výsledků preklinických modelů prokazujících zpomalený zánik motoneuronů díky podpůrné funkci kmenových buněk je v posledních letech zvažována buněčná terapie jako potenciální terapeutická volba. V několika translačních klinických studiích byly testovány různé typy kmenových buněk, stejně jako rozličné aplikační přístupy. Studie se primárně zaměřily na bezpečnost aplikace a druhotně hodnotily vliv kmenových buněk na průběh choroby. Zatímco se buněčná terapie ukázala jako bezpečná, žádná klinická studie dosud neprokázala její pozitivní vliv na průběh choroby. Článek předkládá komentovaný přehled recentních klinických studií s využitím mezenchymálních či neurálních kmenových buněk u pacientů s ALS. Z výsledků studií vyplývá, že pro zhodnocení léčebného efektu kmenových buněk v budoucnu bude zapotřebí jednak standardizovat aplikační postupy a určit nejvhodnějších typ buněk, jednak definovat a validovat spolehlivější biomarkery progrese choroby.
Klíčová slova:
amyotrofická laterální skleróza – buněčná terapie – neurální progenitorové buňky – mezenchymální kmenové buňky – granulocytární kolonie stimulující faktor – klinické studie
Úvod
Amyotrofická laterální skleróza (ALS) je rychle progredující neurodegenerativní onemocnění dospělých, jež primárně postihuje kortikální, bulbární a spinální motoneurony. Příčinou předčasného úmrtí je nejčastěji respirační selhání. Průměrná incidence je v evropské populaci dva případy/ 100 000 obyvatel/rok [1]. Etiologie onemocnění není dosud zcela objasněna [2], ale v posledních letech narůstají díky nově objevovaným kauzálním mutacím důkazy o genetických příčinách nejen u familiárních, ale i u sporadických forem [3]. V histopatologickém nálezu jsou u ~ 96 % všech (familiárních i sporadických) případů ALS prokazovány neuronální a gliové cytoplazmatické inkluze ubikvitinovaného proteinu TDP-43, které mohou být výslednicí předcházejících patofyziologických dějů. Tyto inkluze chybí u případů s prokázanou mutací genu pro superoxid dismutázu-1 (SOD1). Mutace SOD1 představují nejrozšířenější patofyziologický i experimentální terapeutický model choroby, ale reprezentují pouze 1–2 % všech případů ALS. V kontextu absence TDP-43 proteinopatie u SOD1 mutací dosud není objasněno, nakolik lze výstupy z těchto modelů zevšeobecnit na ostatní formy onemocnění [5]. Nejasná patogeneze onemocnění je tedy hlavní příčinou chybějící kauzální terapie. Kromě riluzolu neprokázal žádný léčebný postup významný vliv na průběh nemoci navzdory více než 50 randomizovaným klinickým studiím provedeným během posledních 50 let [6]. Mezi nově zkoušené léčebné postupy patří i buněčná terapie – léčba kmenovými buňkami [7].
Většina experimentálních a tím pádem i translačních studií se opírá o hypotézu, že kmenové buňky (Stem Cells; SC), nejčastěji mezenchymální (Mesenchymal Stem Cells; MSC) nebo neurální (Neural Stem Cells; NSC), jsou schopny podpořit přežití motoneuronů několika různými mechanizmy. Děje se tak prostřednictvím sekrece růstových faktorů [8], imunomodulačním působením na aktivované astrocyty a mikroglii [9], diferenciací do podoby funkční glie [10], resp. více zmiňovanými způsoby současně [11]. Testované způsoby aplikace v preklinických i klinických studiích zahrnují intraspinální, intratékální, intravenózní a nověji i intramuskulární aplikace [12].
Cílem článku je podat komentovaný přehled současných poznatků o výsledcích buněčné terapie v léčbě ALS, které vyplývají z dosud provedených klinických studií.
Metodika a informační zdroje
Studie byly vyhledány v databázi PubMed (http://www.ncbi.nlm.nih.gov/pubmed) pomocí klíčových slov: ALS – amyotrophic lateral sclerosis – stem cells therapy – clinical trials, v období mezi lednem 2009 a lednem 2016 – studie staršího data představují buď dílčí kroky prací recentních nebo testují obdobnou metodiku méně robustně. Studie publikované v neimpaktovaných časopisech nebyly zahrnuty vzhledem k nízké metodologické úrovni. Pomocí těchto kritérií byla nalezena pouze jedna studie, která splňuje kritéria Oxford Centre for Evidence-based Medicine [13] pro minimální kvalitu vědeckého důkazu II, zbývající studie odpovídaly stupni III a IV. Dosud bylo realizováno nejvíce studií s využitím adultních SC vzhledem k nejsnazší dostupnosti těchto linií (MSC, hematopoetické progenitory) a jejich převážně autolognímu původu. Zmiňujeme se i o dvou metodologicky kvalitních pracích s intraspinální aplikací fetálních NSC, které jsou alogenního původu, a při jejich použití je nutná imunosuprese. Studie uvádíme pro přehlednost dle aplikačních přístupů (tab. 1).
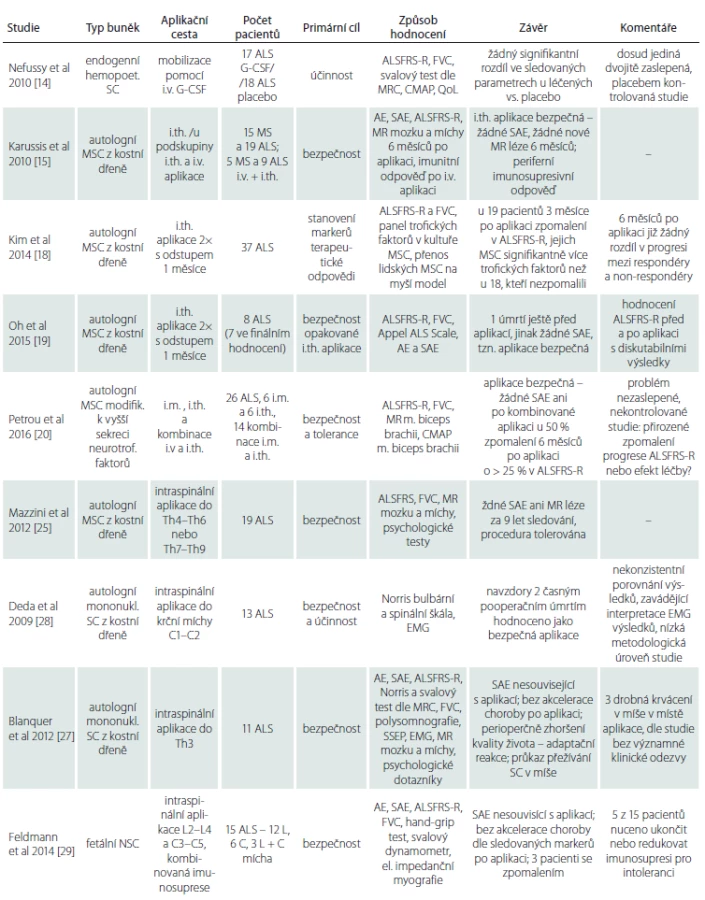

AE – nežádoucí reakce (Adverse Events), ALS – amyotrofická laterální skleróza, ALSFRS-R – revidovaná škála funkčního hodnocení ALS (Revised Amyotrophic Lateral Sclerosis Functional Rat ing Scale), Appel ALS Scale – Appelova škála funkčního zhodnocení ALS, CMAP – sumační svalový akční potenciál (Compound Muscle Action Potential), HŽT – hluboká žilní trombóza, EMG – elektromyografie, FVC – usilovná vitální kapacita (Forced Vital Capacity), G-CSF – faktor stimulující granulocytární kolonie (Granulocyte-Colony Stimulat ing Factor), MS – multiple sclerosis, MSC – mezenchymální kmenové buňky (Mesenchymal Stem Cel ls), MR – magnetická rezonance, MRC – Medical Research Council, MSC – mezenchymové kmenové buňky (Mesenchymal Stem Cel ls), Nor ris bulbární a spinální – Nor risova škála funkčního zhodnocení bulbárních a spinálních symp tomů, NSC – neurální kmenové buňky (Neural Stem Cel ls), QoL – škála kvality života (Quality of Life), SAE – vážné nežádoucí reakce (Serious Adverse Events), SC – kmenové buňky (Stem Cel ls), SSEP – samotosenzorické evokované potenciály.
Systémová – intravenózní aplikace
V roce 2010 publikovali Nefussy et al dvojitě zaslepenou, randomizovanou, placebem kontrolovanou studii s mobilizací autologních hemopoetických progenitorů z kostní dřeně pomocí aplikace faktoru stimulujícího granulocytární kolonie (G-CSF) [14]. Jako primární cíl byl stanoven rozdíl v poklesu anamnestické škály, která hodnotí funkční motorický deficit (Revised ALS Functional Rating Scale; ALSFRS-R). Mezi sekundární cíle patřilo hodnocení poklesu usilovné vitální plicní kapacity (Forced Vital Capacity; FVC), manuálního testu svalové síly dle Medical Research Council (MRC), snížení amplitudy sumačního svalového potenciálu (Compound Muscle Action Potential; CMAP) a hodnocení kvality života. Do studie bylo zařazeno 35 pacientů, z toho 17 pacientů bylo léčeno G-CSF a 18 placebem. Obě skupiny byly srovnatelné věkem, délkou trvání choroby, pohlavím, podílem bulbárních a spinálních forem a vstupním ALSFRS-R skóre. Zvýšení počtu leukocytů a CD34+ buněk prokázalo, že mobilizace autologních progenitorů u léčených pacientů byla dostatečná a léčba byla tolerována bez závažných nežádoucích účinků. Ve srovnání primárních i sekundárních cílů mezi pacienty léčených G-CSF a placebem však žádný signifikantní rozdíl nalezen nebyl.
Intratékální aplikace
V roce 2010 publikovali Karussis et al výsledky studie fáze I zkoumající bezpečnost intratékální (i.th.) aplikace a periferní imunologickou odpověď po intravenózní (i.v.) aplikaci autologních MSC u pacientů s ALS a u pacientů s roztroušenou sklerózou (Multiple Sclerosis; MS) [15]. U 15 MS pacientů a 19 ALS pacientů byla aplikace i.th. a pět MS a devět ALS pacientů obdrželo ještě i.v. aplikaci třetinové dávky. Zhodnocení efektu léčby na průběh choroby nebylo primárním výstupem studie, což odráží i zvolená metodika – heterogenní skupina ALS pacientů dle věku, délky trvání onemocnění a výchozího ALSFRS-R, a velmi strohý rozsah uvedených dat – veškeré údaje, vč. prezentace vývoje ALSFRS-R před aplikací a po ní pouze jako průměrné hodnoty celé skupiny s velkou šíří rozptylu. Klíčové charakteristiky jednotlivých pacientů, které se dle recentních epidemiologických studií ukazují jako determinanty přirozené progrese choroby [16,17], nejsou zřejmé a velikost skupiny je ze statistického pohledu malá. Autoři konstatují stacionární ALSFRS-R po dobu 6 měsíců sledování, což však vzhledem k výše uvedeným metodickým nedostatkům neumožňuje skutečně validní interpretace případného efektu léčby na průběh onemocnění.
V roce 2014 publikovali Kim et al výsledky nezaslepené, nekontrolované studie cílené na detekci potenciálních biomarkerů odpovědi na terapii [18]. Celkem 37 pacientům byly ve dvou i.th. aplikacích s odstupem 1 měsíce podány autologní MSC, čemuž 3 měsíce předcházelo a 6 měsíců následovalo hodnocení ALSFRS-R a FVC. V případě zpomalení rychlosti progrese v ALSFRS-R do 3 měsíců po aplikaci byli pacienti hodnoceni jako respondéři, v opačném případě jako non-respondéři na léčbu. Respondérů bylo 19, non-respondérů 18. Skupiny se statisticky významně nelišily věkem, trváním choroby, zastoupením pohlaví ani rychlostí progrese v ALSFRS-R 3 měsíce před aplikací. Šest měsíců po aplikaci se rychlost progrese u respondérů již signifikantně nelišila od skupiny non-respondérů. Dále byl zhodnocen panel růstových faktorů a interleukinů v kulturách MSC u 13 respondérů a osmi non-respondérů. U tří markerů byla prokázána statisticky významně vyšší střední hladina u respondérů vůči non-respondérům, ovšem s příliš velkým překryvem rozptylů na to, aby bylo možno stanovit cut-off. Souhrnem lze z výsledků této studie usuzovat, že polovina pacientů by neměla z aplikace ani krátkodobý benefit a případný efekt léčby by mohl přetrvat nejdéle 3–6 měsíců. V roce 2015 publikovala stejná skupina studii bezpečnosti opakované i.th. aplikace autologních MSC [19]. Sedm pacientů bylo sledováno dle identického designu jako v předchozí práci. Žádné závažné nežádoucí účinky pozorovány nebyly. Na základě svého designu předkládá studie data týkající se rychlosti progrese pacientů v ALSFRS-R po aplikaci. Tato data je ovšem nutno interpretovat obezřetně. Pro ilustraci: u pacienta s dosavadní progresí – 0,3 bodů/měsíc se rychlost poklesu v ALSFRS-R v tříměsíční preaplikační fázi skokově zvýšila na – 1 bod/měsíc a záhy po aplikaci se na 6 měsíců progrese zcela zastavila. To lze vysvětlit buď náhlou akcelerací choroby v preaplikační fázi, nebo chybou měření. Identický obraz byl u čtyř ze sedmi pacientů, u všech s delším trváním choroby. Hodnoty FVC u těchto pacientů ovšem rychlost poklesu v ALSFRS-R nekopírovaly. Při zhodnocení průměru celého sledovaného období (9 měsíců) byla pak u těchto pacientů rychlost progrese v ALSFRS-R stejná jako před vstupem do studie. Z toho lze usuzovat, že v tříměsíčním období k reálnému významnému zrychlení choroby, a tím pádem ani k následnému zpomalení po aplikaci pravděpodobně nedošlo a jednalo se spíše o variabilitu měření.
V roce 2016 publikovali Petrou et al nezaslepenou nekontrolovanou studii s využitím modifikované linie MSC se zvýšenou sekrecí neurotrofických faktorů [20]. Studie měla dvě fáze, kdy v první byly autologní MSC aplikovány šesti pacientům i.th. a šesti intramuskulárně (i. m.), v navazující druhé fázi pak dalším 14 pacientům kombinovaně oběma přístupy. Primárním cílem studie bylo ověření bezpečnosti a snášenlivosti léčby, sekundárním cílem pak ověření účinku jednak pomocí změny rychlosti progrese v ALSFRS-R, jednak hodnocením objemu svalové hmoty pomocí magnetické rezonance (MR) a změnou poklesu amplitudy CMAP z m. biceps brachii ve srovnání před terapií a po ní. Autoři referují statisticky signifikantní zpomalení rychlosti progrese choroby dle ALSFRS-R (p = 0,052) i v hodnotě FVC (p < 0,04) u pacientů po i.th. nebo kombinované aplikaci. Přes 50 % pacientů dle předkládaných dat vykazovalo zpomalení progrese v ALSFRS-R o více než 25 % proti rychlosti před aplikací, což je dle recentního průzkumu považováno za klinicky dostatečně významnou změnu [21]. Interpretace těchto výsledků je ovšem problematická nejméně ve třech bodech. Prvním z nich je soubor pacientů zařazených do studie: pravděpodobná či definitivní ALS dle revidovaných El Escorial kriterií, tzv. klasická forma ALS a čas od prvních příznaků méně než 2 roky. Právě u pacientů s těmito charakteristikami totiž dochází dle analýzy přirozeného průběhu nemoci ve druhém roce od diagnózy ke zpomalení rychlosti progrese v ALSFRS-R [17]. Problematický je i model srovnání předpokládané rychlosti progrese vůči skutečné, po aplikaci pozorované, rychlosti progrese v této škále, pomocí kterého je dokládáno zpomalení po aplikaci. Odhad předpokládané rychlosti vychází totiž z prosté lineární regrese dat úvodních 3 měsíců před aplikací. Lineární regrese je dle recentních prací ovšem zatížena významnou odchylkou a přesnější odhad poskytují exponenciální modely odhadu [16,22]. Nakonec dle analýzy přirozeného průběhu u více než 3 000 pacientů jich během šestiměsíčního sledování 25 % v ALSFRS-R neprogredovalo [23]. Výše zmíněné poznatky ilustrují potřebu placebem kontrolované, dvojitě slepé studie, stejně jako spolehlivějších a objektivnějších markerů progrese. V této studii sledovaný longitudinální pokles amplitudy CMAP m. biceps brachii v čase vykazoval dle autorů po i. m. aplikaci statisticky nesignifikantní tendenci k zpomalení poklesu. Amplituda CMAP ovšem není dostatečně validovaný biomarker a jeho limitaci představuje proces přirozené reinervace ze zachovalých motoneuronů [24]. Změna objemu m. biceps brachii po i. m. aplikaci nebyla navíc dle MR hodnocení významná proti neléčené straně.
Intraspinální aplikace
V roce 2012 publikovali Mazzini et al souhrnné výsledky devítiletého sledování, kdy byly celkem 19 pacientům intraspinálně aplikovány autologní MSC [25]. Tyto studie nelze nazvat translační, neboť zvířecí model byl testován ex post [7]. Hlavním cílem studie byla monitorace bezpečnosti a vhodnosti této aplikační cesty. Pacientům bylo po šestiměsíčním úvodním sledování operačně aplikováno do hrudní míchy různé množství autologních MSC. Následoval monitoring pomocí MR míchy, klinického a psychologického zhodnocení po 3 měsících až do smrti pacienta. Ve výsledku nebyly zaznamenány žádné závažné nežádoucí účinky, avšak ani žádný významný klinický přínos z aplikace. Zpomalení progrese, pozorované u šesti pacientů, interpretují autoři spíše jako přirozený průběh choroby – jednalo se o nejmladší z pacientů, resp. o pacienty s izolovaným postižením dolního motoneuronu s nejpomalejším průběhem choroby. Dlouhodobé psychologické monitorování robustní baterií dotazníků ukázalo, že majoritní podíl na vnímané kvalitě života má především sociální zázemí, stejně jako premorbidně aktivní postoj k životu. To je v souladu se zjištěním jiných studií, které hodnotily determinanty kvality života a adaptaci na chorobu ALS [26]. Ve výše uvedeném sledování se subjektivně vnímaná kvalita života navzdory invazivní proceduře nezhoršila.
V roce 2012 publikovali Blanquer et al výsledky intraspinální aplikace autologních mononukleárních buněk z kostní dřeně 11 pacientům [27]. Výsledky s aplikací této buněčné linie publikovali Deda et al již v roce 2009. Studie ovšem měla nízkou metodologickou kvalitu, a proto není v našem článku více komentována [28]. Design studie Blanquera et al byl cílen na ověření bezpečnosti, proto byla zvolena relativně bezpečná aplikace z laminektomie Th3–Th4. Aplikaci půl roku předcházelo a 1 rok následovalo robustní monitorování klinické, spirometrické, MR mozku a míchy, elektrofyziologické a psychologické kontroly v tříměsíčních intervalech. V rámci průměrných hodnot celé skupiny nedošlo po aplikaci k významnému urychlení progrese ve spirometrii a v ALSFRS-R. Psychologické hodnocení ukázalo pokles kvality života v období posledního měsíce před aplikací, což autoři interpretovali jako adaptační reakci. V pooperačním období se pak většina pacientů stran kvality života stabilizovala na hodnotě, se kterou do studie vstupovali. U čtyř pacientů, kteří v důsledku progrese nemoci zemřeli, bylo pak provedeno histologické a histochemické zhodnocení míchy. Zde autoři dokládají signifikantně větší počet motoneuronů v míšních segmentech v místě aplikace proti neaplikovaným míšním segmentům u tří pacientů a zároveň dlouhodobé přežívání graftu. V závěru této metodologicky kvalitní studie autoři konstatují, že je nutné bližší pochopení biologických mechanizmů stojících v pozadí buněčné terapie.
V roce 2014 publikovali Feldmanová et al souhrnné výsledky studie na 15 pacientech s ALS, kdy byla pacientům operačně z laminektomie za pomocí speciálního mikroinjektoru intraspinálně aplikována linie alogenních neurálních progenitorů z fetální míchy [29]. Cílem bylo zhodnotit bezpečnost tohoto postupu. Pacienti byli rozděleni do pěti kohort jednak dle principu eskalace dávky (od uni- přes bilaterální lumbální až ke kombinované dvoudobé lumbální a cervikální aplikaci), jednak dle principu eskalace rizika (od nejvíce postižených k těm s malým deficitem). K prevenci rejekce byla podávána imunosuprese ve schématu jako u orgánové transplantace. Fáze před aplikací a po ní byla zhodnocena pomocí ALSFRS-R, FVC a baterií dalších biomarkerů (např. handgrip test, svalový dynamometr, elektrická impedanční myografie). Porovnání rychlostí progrese mělo vyloučit akceleraci progrese choroby po aplikaci. Problematická byla udržovací imunosuprese, kterou z důvodů gastrointestinální intolerance ukončilo či redukovalo pět z 15 pacientů. Ke zrychlení progrese choroby, dle použitých biomarkerů, nedošlo u žádného pacienta a u tří došlo k jejímu zpomalení, u jednoho dokonce k přechodnému zlepšení. Dva z těchto tří pacientů byli ve studii nejmladší a s nejkratším trváním choroby. Efekt zpomalení progrese choroby postupně klesal s časem od aplikace a u dvou pacientů z kohorty s dvoudobým výkonem po aplikaci do krční míchy opět vzrostl. To autoři interpretovali jako určitou tendenci k přínosu opakovaného podání, stejně jako víceetážové aplikace.
V roce 2015 publikovali Mazzini et al iniciální výsledky bezpečnostní studie s intraspinální aplikací fetálních NSC šesti imobilním pacientům v pokročilém stadiu choroby [30]. Tři pacienti obdrželi unilaterální, tři bilaterální aplikaci buněk do lumbální intumescence, opět na principu eskalace dávky. Byl využit stejný stereotaktický mikroinjektor jako v předešlé studii, ovšem s redukovaným schématem imunosuprese (pouze takrolimus, ukončen po půl roce). Sledování a škálování probíhalo každý měsíc po dobu 1 roku, poté každé 3 měsíce až do smrti. MR kontroly mozku a míchy po výkonu probíhaly čtvrtletně. Byl použit robustní panel testování kvality buněk a zhodnocení jejich tumorigenního potenciálu, vč. in vivo testu na imunodeficientních myších. Kromě jedné flebotrombózy se nevyskytly závažné nežádoucí reakce a zároveň pacienti lépe tolerovali imunosupresi proti předchozí studii. MR kontroly byly bez signifikantních strukturálních změn. Během sledování nedošlo k akceleraci progrese choroby, významný efekt léčby však zaznamenán nebyl. Souhrnem autoři konstatují, že jejich hlavním cílem bylo co nejvíce standardizovat intraspinální postup aplikace NSC, testovat jejich kvalitu a dále reprodukovat metodu Feldmannové et al, se kterými hodlají vytvořit multicentrickou studii.
Intracerebrální aplikace
V roce 2009 publikovali Martinez et al nezaslepenou, nerandomizovanou studii se stereotaktickou aplikací MSC do frontálního kortexu [31]. Bylo porovnáno 10 léčených a 13 neléčených pacientů. Ve studii byl dokládán efekt léčby třikrát delším průměrným přežitím léčené skupiny. Ovšem tato skupina měla před vstupem do studie dvakrát delší dobu trvání choroby než kontroly, a jednalo se tedy spíše o pacienty s pomalu progredující chorobou. Tvrzení, že se někteří pacienti po 6 měsících od léčby zlepšili až o 13 bodů v ALSFRS-R, působí nevěrohodně v situaci, kdy aplikace byla provedena pouze z jednoho vpichu do motorické arey každé strany.
Shrnutí a perspektivy
Ačkoliv mnohé experimentální práce dokládají pozitivní efekt buněčné terapie na průběh nemocí motoneuronů, dosud publikované klinické studie tento efekt zatím nepotvrzují. Důvodem je v první řadě skutečnost, že k tomu buď nebyly adekvátně navrženy – primárním cílem studií bylo spíše hodnocení bezpečnosti léčby a způsobu aplikace než vliv léčby na průběh nemoci, nebo v důsledku svých metodologických limitací, jak bylo nastíněno v komentářích k jednotlivým studiím. Problém translace nadějných výsledků z myších SOD1 modelů do úspěšných klinických studií je obecně pozorovaným jevem všech dosud testovaných léčiv u ALS [6,32]. Existuje několik faktorů, kterými lze tento fenomén vysvětlit. Prvním faktorem je, že v drtivé většině experimentálních studií je léčba zahájena v presymptomatické fázi nebo bezprostředně po rozvoji příznaků [32]. Vzhledem k průměrné prodlevě 11 měsíců od prvních symptomů k diagnóze [33] nelze tuto situaci v klinických studiích reprodukovat a v kontextu rychle progredující neurodegenerace se jedná o výrazně pokročilejší patologický proces. Druhým faktorem, který se dotýká preklinických prací, je publikační bias neboli tendence častěji publikovat výsledky prokazující pozitivní účinek zkoumaného terapeutického postupu a naopak negativní výsledky nepublikovat, což poté zkresluje obraz počtu úspěšných experimentálních studií [32,34]. Třetím faktorem je, že mnoho experimentálních studií nesplňuje přísné metodologické požadavky kladené na studie klinické v podobě zaslepení či randomizace, což se odráží v podobě selekčních či detekčních bias a podmiňuje tendence k pozitivnímu nadhodnocení výsledků [34]. Byly vypracovány standardní doporučené postupy, na jejichž podkladě lze, obdobně jako u studií klinických, zhodnotit metodologickou úroveň experimentálních studií [35]. Posledním faktorem, který může znesnadnit translaci výsledků experimentálních studií, je nejasná relevance výstupů z modelů SOD1 mutací pro ostatní formy ALS [6].
Má-li buněčná terapie v budoucnosti prokázat svoji účinnost v léčbě ALS, bude potřeba splnit více požadavků ještě na preklinické úrovni. V situaci, kdy jednotlivé experimentální studie pracují s velkým množstvím různých buněčných linií s odlišnými vlastnostmi, konkrétně odlišnou mírou neurotropizmu, sekrece růstových faktorů, imunomodulačních vlastností, nebo odlišným původem (auto- vs. alogenní), bude jednou z prvních priorit identifikace terapeuticky nejefektivnějšího typu buněk a následně standardizování jeho vlastností [36]. Autologní MSC sice nevyžadují imunosupresi, ale jejich nevýhodou je jednak experimentálně prokázaný vliv senescence, tedy poklesu funkčních schopností MSC s narůstajícím věkem donora [37], jednak jejich významně horší funkční vlastnosti v korelaci s aktivitou ALS [38], resp. v důsledku přítomností kauzální mutace [39]. Na druhé straně pak experimentální studie na myším modelu sice prokazují významně delší přežívání instraspinálního graftu humánních NSC díky kombinované imunosupresi (takrolimus a mykofenolát mofetil) [40], avšak v klinické studii Feldmannové et al musela až třetina pacientů tuto terapii ukončit či významně redukovat pro intoleranci [29].
Dále je potřeba definovat a standardizovat aplikační metody zajišťující optimální průnik dostatečného množství buněk do všech míst postižených patologií. Tuto problematiku ilustrují experimentální modely intraspinální aplikace NSC, které sice prokazují zvýšený počet přežívajících motoneuronů v blízkosti místa aplikace, ovšem tento efekt mizí se vzdáleností a stejně tak nedochází k žádnému pozitivnímu ovlivnění kortikálního motoneuronu a descendentních drah, a tím pádem ani k podstatnému a trvalejšímu funkčnímu zlepšení [41]. Tyto experimentální výsledky tak dále objasňují limitovaný efekt jednoetážové intraspinální aplikace v publikovaných klinických studiích. Benefit i.th. aplikace je na technické i bezpečnostní rovině nesporný, výsledky i.th. aplikací (humánních) MSC myším modelům ovšem svědčí o velmi omezenému průniku těchto MSC do parenchymu. Pozorovaný efekt na délku přežití a zlepšení funkčních parametrů je tak přisuzován nejspíše jejich parakrinnímu působení z likvorového rezervoáru a zároveň vyžaduje opakovanou aplikaci [42]. Je však nezbytné určit vhodné intervaly opakování aplikací, stejně jako vhodné dávky, o což se již pokusily některé experimentální [42,43] a v omezené míře i klinické práce [18,20,27].
Provedené klinické studie s intraspinální aplikací SC přinášejí většinou důkazy o relativní bezpečnosti tohoto přístupu, dalším klíčovým krokem se jeví standardizace jednak operačních protokolů, jak ukazují recentní práce Feldmannové a Mazziniové, jednak i pooperační péče. Nadále bude potřeba prokázat v širším rozsahu, že skutečně nedochází k pooperační akceleraci choroby, čemuž naopak nasvědčují observační studie na nepoměrně větších souborech pacientů [44]. Pro bezpečnost autologních kmenových buněk v klinické aplikaci svědčí provedené metaanalýzy [45], pro humánní aplikaci NSC metaanalýzy dosud chybí a kazuistiky nasvědčují riziku maligní transformace u imunosuprimovaných jedinců [46], která je právě u tohoto buněčného typu relevantní. Pro testování bezpečnosti bude nutné vytvořit standardní postup např. dle modelu poslední studie Mazziniho et al [30]. Hodnocení klinického efektu terapie bude vyžadovat spolehlivější objektivní biomarkery progrese nemoci a odpovědi na léčbu, které se vyhnou stávajícím limitacím všeobecně užívané škály ALSFRS-R, zejména její nízké senzitivitě [24]. Podle novějších poznatků se jako vhodná jeví revize a další upřesnění této škály [47]. Panel případných biomarkerů by mohl umožnit definovat a charakterizovat pacienty odpovídající na léčbu, jak se o to pokusila jedna z referovaných studií [18]. Předběžné poznatky z citovaných klinických studií nastolují také otázku o přínosu buněčné terapie v závislosti na délce trvání choroby a stupni klinického postižení ve smyslu hranice, po jejímž překročení už nelze očekávat významný příznivý léčebný efekt. Pro design budoucích klinických studií se dále jako klíčová jeví detailnější stratifikace pacientů do skupin rychle a pomalu progredujících [22,48], např. dle recentních prognostických modelů zohledňujících i přítomnost kognitivního deficitu dysexekutivního typu jako významného rizikového faktoru rychleji progredující formy ALS [49]. V kontextu odhadů, podle kterých je potřeba sledovat 170 pacientů s rychle progredující formou ALS nejméně 12 měsíců, aby bylo možné s dostatečnou statistickou silou určit 20% zpomalení progrese dle škály ALSFRS-R [22], je zřejmé, že studie k průkazu účinku terapie by měly probíhat jednak na multicentrické úrovni k zajištění dostatečného počtu účastníků, jednak nejméně rok. Ve složité otázce financování se při nedostupnosti či nedostatečném rozsahu standardních grantových prostředků jako viabilní alternativa jeví např. metoda tzv. crowdfundingu [50], která se vyhýbá etickým úskalím metod spojených se spoluúčastí pacientů samotných.
Seznam použitých zkratek
ALS –amyotrofická laterální skleróza (Amyotrophic Lateral Sclerosis)
ALSFRS-R – revidovaná škála funkčního hodnocení ALS (Revised Amyotrophic Lateral Sclerosis Functional Rating Scale)
CMAP – amplituda sumačního svalového potenciálu (Compound Muscle Action Potential)
FVC – usilovná vitální kapacita (Forced Vital Capacity)
G-CSF – faktor stimulující granulocytární kolonie (Granulocyte-Colony Stimulating Factor)
MS – roztroušená skleróza (Multiple Sclerosis)
MR – magnetická rezonance
MSC – mezenchymální kmenové buňky (Mesenchymal Stem Cells)
NSC – neurální kmenové buňky (Neural Stem Cells)
SOD1 – superoxid dismutáza 1
TDP-43 – Transactive DNA binding Protein of 43 kDa length
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Daniel Baumgartner
Neurologická klinika
2. LF UK a FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: daniel.baumgartner@fnmotol.cz
Přijato k recenzi: 4. 4. 2016
Přijato do tisku: 27. 7. 2016
Zdroje
1. Chiò A, Logroscino G,Traynor BJ, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 2013;41(2):118– 30. doi: 10.1159/ 000351153.
2. Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013; 14(4):248– 64. doi: 10.1038/ nrn3430.
3. Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014;17(1):17– 23. doi: 10.1038/ nn.3584.
4. Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA--binding protein 43 in neurodegenerative disease. Nat Rev Neurol 2010;6(4):211– 20. doi: 10.1038/ nrneurol.2010.18.
5. Turner MR, Hardiman O, Benatar M, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol 2013;12(3):310– 22. doi: 10.1016/ S1474-4422(13)70036-X.
6. Mitsumoto H, Brook BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol 2014;13(11):1127– 38. doi: 10.1016/ S1474-4422(14)70129-2.
7. Gordon P, Corcia P, Meininger V. New therapy options for amyotrophic lateral sclerosis. Expert Opin Pharmacother 2013;14(14):1907– 17. doi: 10.1517/ 14656566.2013.819344.
8. Hwang DH, Lee HJ, Park IH, et al. Intrathecal transplantation of human neural stem cells overexpressing VEGF provide behavioral improvement, disease onset delay and survival extension in transgenic ALS mice. Gene Ther 2009;16(10):1234– 44. doi: 10.1038/ gt.2009.80.
9. Vercelli A, Mereuta OM, Garbossa D, et al. Human mesenchymal stem cell transplantation extends survival, improves motor performance and decreases neuroinflammation in mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 2008;31(3):395– 405. doi: 10.1016/ j.nbd.2008.05.016.
10. Lepore AC, Rauck B, Dejea C, et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nature Neurosci 2008;11(11):1294– 301. doi: 10.1038/ nn.2210.
11. Teng YD, Benn SC, Kalkanis SN, et al. Multimodal actions of neural stem cells in a mouse model of ALS: a meta-analysis. Sci Transl Med 2012;4(165):165ra164. doi: 10.1126/ scitranslmed.3004579.
12. Goutman SA, Chen KS, Feldman EL. Recent advances and the future of stem cell therapies in amyotrophic lateral sclerosis. Neurotherapeutics 2015;12(2):428– 48. doi: 10.1007/ s13311-015-0339-9.
13. OCEBM Levels of Evidence Working Group. The Oxford 2011 Levels of Evidence. Oxford Centre for Evidence-Based Medicine. [online]. Available from URL: http://www.cebm.net/index.aspx?o=5653.
14. Nefussy B, Artamonov I, Deutsch V, et al. Recombinant human granulocyte-colony stimulating factor administration for treating amyotrophic lateral sclerosis: a pilot study. Amyotroph Lateral Scler 2010;11(1– 2):187– 93. doi: 10.3109/17482960902933809.
15. Karussis D, Karageorgiou C, Vaknin-Dembinsky A, et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol 2010;67(10):1187– 94. doi: 10.1001/ archneurol.2010.248.
16. Gordon PH, Cheng B, Salachas F, et al. Progression in ALS is not linear but is curvilinear. J Neurol 2010;257(10):1713– 7. doi: 10.1007/ s00415-010-5609-1.
17. Mandrioli J, Biguzzi S, Guidi C, et al. Heterogeneity in ALSFRS-R decline and survival: a population based study in Italy. Neurol Sci 2015;36(12):2243– 52. doi: 10.1007/s10072-015-2343-6.
18. Kim HY, Kim H, Oh KW, et al. Biological markers of mesenchymal stromal cells as predictors of response to autologous stem celltransplantation in patients with amyotrophic lateral sclerosis: an investigator-initiated trial and in vivo study. Stem Cells 2014;32(10):2724– 31. doi: 10.1002 / stem.1770.
19. Oh KW, Moon C, Kim HY, et al. Phase I trial of repeated intrathecal autologous bone marrow-derived mesenchymal stromal cells in amyotrophiclateral sclerosis. Stem Cells Transl Med 2015;4(6):590– 7. doi: 10.5966/sctm.2014-0212.
20. Petrou P, Gothelf Y, Argov Z, et al. Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation inp with amyotrophic lateral sclerosis: results of phase 1/ 2 and 2a clinical trials. JAMA Neurol 2016;73(3):337– 44. doi: 10.1001/ jamaneurol.2015.4321.
21. Castrillo-Viguera C, Grasso DL, Simpson E. Clinical significance in the change of decline in ALSFRS-R. Amyotroph Lateral Scler 2010;11(1– 2):178– 80. doi: 10.3109/ 17482960903093710.
22. Gomeni R, Fava M, PRO-ACT Database. Amyotrophic lateral sclerosis disease progression model. Amyotroph Lateral Scler Frontotemporal Degener 2014;15:119– 29. doi: 10.3109/ 21678421.2013.838970.
23. Bedlack RS, Vaughan T, Wicks P, et al. How common are ALS plateaus and reversals? Neurology 2016;86(9):808– 12. doi: 10.1212/ WNL.0000000000002251.
24. Simon NG, Turner MR, Vucic S, et al. Quantifying disease progression in amyotrophic lateral sclerosis. Ann Neurol 2014;76(5):643– 57. doi: 10.1002/ ana.24273.
25. Mazzini L, Mareschi K, Ferrero I, et al. Mesenchymal stromal cell transplantation in amyotrophic lateral sclerosis: a long-term safety study. Cytotherapy 2012;14(1):56– 60. doi: 10.3109/ 14653249.2011.613929.
26. Matuz T, Birbaumer N, Hautzinger M, et al. Coping with amyotrophic lateral sclerosis: an integrative view. J Neurol Neurosurg Psychiatry 2010;81(8):893– 8. doi: 10.1136/ jnnp.2009.201285.
27. Blanquer M, Moraleda JM, Iniesta F, et al. Neurotrophic bone marrow cellular nests prevent spinal motoneuron degeneration in amyotrophic lateral sclerosis patients: a pilot safety study. Stem Cells 2012;30(6):1277– 85. doi: 10.1002/ stem.1080.
28. Deda H, Inci MC, Kürekçi AE, et al. Treatment of amyotrophic lateral sclerosis patients, by autologous bone marrow-derived hematopoietic stem cell transplantation: a 1-year follow-up. Cytotherapy 2009;11(1):18– 25. doi: 10.1080/ 14653240802549470.
29. Feldman EL, Boulis NM, Hur J, et al. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann Neurol 2014;75(3):363– 73. doi: 10.1002/ ana.24113.
30. Mazzini L, Gelati M, Profico DC, et al. Human neural stem cell transplantation in ALS: initial results from a phase I trial. J Transl Med 2015;13:17. doi: 10.1186/ s12967-014-0371-2.
31. Martinez HR, Gonzalez-Garza MT, Moreno-Cuevas JE, et al. Stem-cell transplantation into the frontal motor cortex in amyotrophic lateral sclerosis patients. Cytotherapy 2009;11(1):26– 34. doi: 10.1080/ 14653240802644651.
32. Benatar M. Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol Dis 2007;26(1):1– 13. doi: 10.1016/ j.nbd.2006.12.015.
33. Chiò A, Calvo A, Moglia C, et al. Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry 2011;82(7):740– 6. doi: 10.1136/ jnnp.2010.235952.
34. van der Worp HB, Howells DW, Sena ES, et al. Can animal models of disease reliably inform human studies? PLoS Med 2010;7(3):e1000245. doi: 10.1371/ journal.pmed.1000245.
35. Ludolph AC, Bendotti C, Blaugrund E, et al. Guidelines for preclinical animal research in ALS/ MND: a consensus meeting. Amyotroph Lateral Scler 2010;11(1– 2):38– 45. doi: 10.3109/ 17482960903545334.
36. Coatti GC, Beccari MS, Olávio TR, et al. Stem cellsfor amyotrophic lateral sclerosis modeling and therapy: myth or fact? Cytometry A 2015;87(3):197– 211. doi: 10.1002/ cyto.a.22630.
37. Scruggs BA, Semon JA, Zhang X, et al. Age of the donor reduces the ability of human adipose-derived stem cells to alleviate symptoms in the experimental autoimmune encephalomyelitis mouse model. Stem Cells Transl Med 2013;2(10):797– 807. doi: 10.5966/ sctm.2013-0026.
38. Koh SH, Baik W, Noh MY, et al. The functional deficiency of bone marrow mesenchymal stromal cells in ALS patients is proportional to disease progression rate. Exp Neurol 2012;233(1):472– 80. doi: 10.1016/ j.expneurol.2011.11.021.
39. Boucherie C, Caumont AS, Maloteaux JM, et al. In vitro evidence for impaired neuroprotective capacities of adult mesenchymal stem cells derived from a rat model of familial amyotrophic lateral sclerosis (hSOD1(G93A)). Exp Neurol 2008;212(2):557– 61. doi: 10.1016/ j.expneurol.2008.04.030.
40. Hefferan MP, Johe K, Hazel T, et al. Optimization of immunosuppressive therapy for spinal grafting of human spinal stem cells in a rat model of ALS. Cell Transplant 2011;20(8):1153– 61. doi: 10.3727/ 096368910X564553.
41. Hefferan MP, Galik J, Kakinohana O, et al. Human neural stem cell replacement therapy for amyotrophic lateral sclerosis by spinal transplantation. PLoS One 2012;7(8):e42614. doi: 10.1371/ journal.pone.0042614.
42. Zhang C, Zhou C, Teng JJ, et al. Multiple administrations of human marrow stromal cells through cerebrospinal fluid prolong survival in a transgenic mouse model of amyotrophic lateral sclerosis. Cytotherapy 2009;11(3):299– 306. doi: 10.1080 / 14653240902806986.
43. Kim H, Kim HY, Choi MR, et al. Dose-dependent efficacy of ALS-human mesenchymal stem cells transplantation into cisterna magna in SOD1-G93A ALS mice.Neurosci Lett 2010;468(3):190– 4. doi: 10.1016/ j.neulet.2009.10.074.
44. Pinto S, Swash M, de Carvalho M. Does surgery accelerate progression of amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry 2014;85(6):643– 6. doi: 10.1136/ jnnp-2013-305770.
45. Lalu MM, McIntyre L, Pugliese C, et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials. PLoS One 2012;7(10):e47559. doi: 10.1371/ journal.pone.0047559.
46. Amariglio N, Hirshberg A, Scheithauer BW, et al. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med 2009;6(2):e1000029. doi: 10.1371/ journal.pmed.1000029.
47. Franchignoni F, Mora G, Giordano A, et al. Evidence of multidimensionality in the ALSFRS-R Scale: a critical appraisal on its measurement properties using Rasch analysis. J Neurol Neurosurg Psychiatry 2013;84(12):1340– 5. doi: 10.1136/ jnnp-2012-304701.
48. Proudfoot M, Jones A, Talbot K, et al. The ALSFRS as an outcome measure in therapeutic trials and its relationship to symptom onset. Amyotroph Lateral Scler Frontotemporal Degener 2016;17(5– 6):414– 25. doi: 10.3109/ 21678421.2016.1140786.
49. Elamin M, Bede P, Montuschi A, et al. Predicting prognosis in amyotrophic lateral sclerosis: a simple algorithm. J Neurol 2015;262(6):1447– 54. doi: 10.1007/ s00415-015-7731-6.
50. Sharma A, Khan JS, Devereaux PJ. Is crowdfunding a viable source of clinical trial research funding? Lancet 2015; 386(9991):338. doi: 10.1016/ S0140-6736(15)61407-6.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2017 Číslo 1
Nejčtenější v tomto čísle
- Není třeba léčit motorické tiky
- Základní neurologické vyšetření – nastal čas pro změny?
- Současný pohled na kontraindikace a komplikace elektromyografie
- Endoskopická exstirpace koloidní cysty III. mozkové komory