
Význam nových laboratorních technik v diagnostice Niemann-Pickovy choroby typu C
Role of novel laboratory techniques in Niemann-Pick type C disease diagnostics
This review provides a summary of current approaches to Niemann-Pick disease type C (NP-C) diag-nostics with an emphasis on novel laboratory techniques. NP-C is a severe autosomal recessive neurovisceral disorder and the recent availability of disease-modifying therapies increases the importance of its timely diagnosis. The hereditary deficiency of cholesterol transporter proteins (NPC1 or NPC2) in NP-C leads to abnormal intracellular lipid trafficking. Clinical suspicion for NP-C has to be confirmed by biochemical and/or molecular genetic methods. Novel biomarkers in serum or plasma and advanced sequencing techniques now have a prominent role in NP-C diagnostics. In a subset of patients, it is necessary to use several complementary techniques for confirmation of NP-C diagnosis, including advanced biochemical and cellular assays discussed in the paper. These methods therefore have to be available in a specialized laboratory.Niemann-Pickova choroba typu C – diagnostika – intracelulární transport cholesterolu – biomarkery – lyzosfi ngolipidy – oxysteroly – fi lipinový test
Keywords:
biomarkers – oxysterols – Niemann-Pick disease type C – diagnostics – intracellular cholesterol traffi cking – lysosphingolipids – filipin test
Authors:
M. Hřebíček 1; H. Jahnová 2; L. Dvořáková 1,2; F. Majer 1; D. Mušálková 1; J. Ledvinová 1; L. Kuchař 1
Authors‘ workplace:
Laboratoř pro studium vzácných nemocí, Klinika dětského a dorostového, lékařství 1. LF UK a VFN v Praze
1; Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
2
Published in:
Cesk Slov Neurol N 2020; 83/116(3): 263-268
Category:
Review Article
doi:
https://doi.org/10.14735/amcsnn2020263
Overview
Cílem sdělení je shrnutí současného přístupu k diagnostice Niemann-Pickovy choroby typu C (NP-C) s důrazem na nové laboratorní techniky. NP-C je závažné autozomálně recesivní neuroviscerální onemocnění a aktuální dostupnost terapie ovlivňující průběh nemoci zvyšuje důležitost její včasné diagnostiky. Dědičně podmíněný deficit transportního proteinu pro cholesterol (NPC1 nebo NPC2) u NP-C vede k poruše transportu lipidů uvnitř buňky. Ověření klinického podezření na NP-C se proto opírá o biochemické a/nebo molekulárně genetické metody. Nové metody využívajících analýzu biomarkerů v krevním séru nebo plazmě a pokročilé sekvenační techniky mají nyní v diagnostice NP-C důležitou roli. U části pacientů je pro ověření diagnózy nutné použít více vzájemně se doplňujících vyšetření, vč. v článku diskutovaných pokročilých buněčných a biochemických technik. Ty proto musí být k disposici ve specializované laboratoři.
Klíčová slova:
Niemann-Pickova choroba typu C – diagnostika – intracelulární transport cholesterolu – biomarkery – lyzosfi ngolipidy – oxysteroly – filipinový test
Úvod
Výzkum Niemann-Pickovy choroby typu C (NP-C) vedl v nedávné době k zavedení nových diagnostických technik a k vývoji slibných terapeutických postupů. Rovněž se rozšířily naše znalosti o fenotypovém spektru onemocnění, zejména o dospělých formách nemoci. Cílem tohoto článku je informovat o nových metodách a shrnout, jak se tyto změny odrážejí v současném diagnostickém přístupu k NP-C.
Niemann-Pickova choroba typu C
Niemann-Pickova choroba typu C je závažné autozomálně recesivní neuroviscerální metabolické onemocnění, které se celosvětově vyskytuje s frekvencí zhruba 1: 100 000 živě narozených. Typické střádání neesterifikovaného cholesterolu, v menší míře i sfingomyelinu a některých glykolipidů v lysozomech a pozdních endozomech, je způsobeno deficity transportních proteinů NPC1 nebo NPC2. Celosvětově jsou zhruba u 95 % pacientů s NP-C nalezeny mutace v genu NPC1, u zbývajících případů je mutován gen NPC2. Oba geny kódují transportní proteiny pro cholesterol v pozdně endozomálním/lysozomálním kompartmentu (LE/LY) [1].
Klinický obraz onemocnění
Klinický obraz NP-C je velmi variabilní a obvykle zahrnuje neurologické a viscerální příznaky (postižení jater, sleziny a někdy i plic), které se typicky objevují dříve nebo současně s neurologickými. Věk rozvoje neurologických a psychiatrických příznaků je však rozhodující pro klinickou klasifikaci většiny forem NP-C (časně infantilní, pozdně infantilní, juvenilní, adolescentní/adultní). Klinický obraz NP-C je předmětem řady přehledných článků, na které čtenáře proto odkazujeme – článek Jahnové et al [2] a další literaturu [1,3,4]. Pro identifikaci pacientů s podezřením na NP-C lze také využít klinické skórování [5] s upravenou alternativou pro pacienty mladší 4 let [6].
Adultní/adolescentní forma NP-C je dnes považována za nejčastější, následovaná juvenilní formou. V ČR v posledních 15 letech odpovídá zastoupení nově diagnostikovaných pacientů s adolescentní/adultní formou NP-C 40–50 % (vlastní soubor). Přesto nelze vyloučit, že řada dospělých pacientů stále uniká diagnóze.
Miglustat (Zavesca, Actelion Pharmaceutials, Allschwil, Švýcarsko), inhibitor syntézy glykosfingolipidů, je jediným preparátem schváleným pro léčbu NP-C. Výsledky dlouhodobého multicentrického sledování pacientů na léčbě svědčí pro skutečnost, že preparát zpomaluje průběh NP-C [7]. V klinickém zkoušení je intratékálně aplikovaný 2-hydroxypropyl-b-cyklodextrin [8], který ne zcela objasněným mechanizmem překonává blok transportu cholestrolu v LE/LY, a arimoclomol, který indukuje HSP70 a další heat shock proteiny, jejichž zvýšená exprese snižuje lysozomální střádání u modelů několika lysozomálních onemocnění vč. NP-C [9].
Intracelulární metabolizmus cholesterolu a NP-C
Niemann-Pickova choroba typu C je poruchou nitrobuněčného transportu lipidů, především neesterifikovaného cholesterolu pocházejícího z lipoproteinů o nízké hustotě (low density lipoprotein; LDL). LDL jsou významným zdrojem cholesterolu pro buňky. Jsou endocytované prostřednictvím LDL-receptorů [10]. V pozdních endozomech jsou estery cholesterolu z LDL hydrolyzovány kyselou lipázou, která uvolní z esterové vazby neesterifikovaný cholesterol (obr. 1). Ten je navázán na luminální transportní protein NPC2, který jej předá transmembránovému transportéru NPC1. NPC1 přenese cholesterol přes glykokalyx, oligosacharidovou vrstvu na vnitřní straně lysozomální membrány a uvolní ho do lipidové dvojvrstvy membrány [11]. Odtud je cholesterol transportován do dalších membránových kompartmentů (endoplazmatické retikulum, mitochondrie, plazmatická membrána, peroxizomy). Při tom se uplatňuje také transport prostřednictvím membránových kontaktních míst (membrane contact site; MCS) dvou subcelulárních kompartmentů [12]. Řadu aspektů intracelulárního transportu cholesterolu však stále neznáme. Deficit NPC1 nebo NPC2 vede k hromadění cholesterolu, sfingomyelinu a dalších lipidů v LE/LY a je příčinou NP-C [13]. Akumulace cholesterolu v LE/LY vede k oxidativnímu stresu, zvýšené tvorbě kyslíkových radikálů a oxysterolů, oxidativních produktů cholesterolu [14].
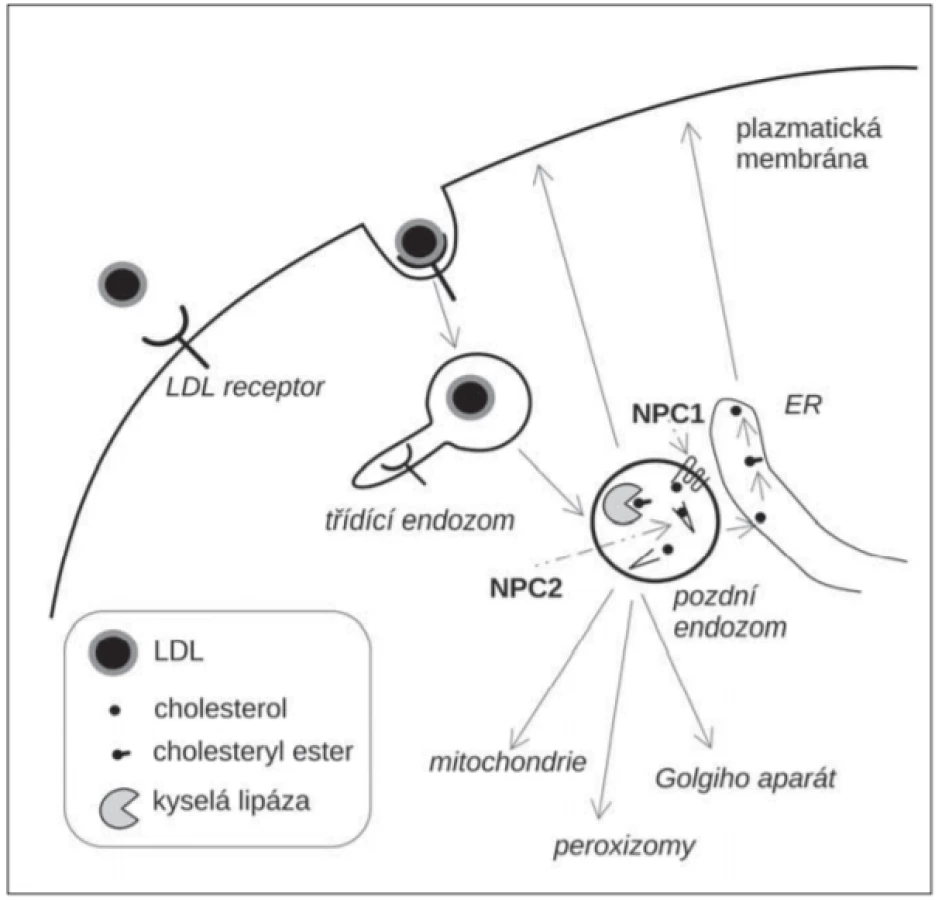
Fig. 1. A schematic representation of intracellular traffi cking of LDL-cholesterol.
ER – endoplasmic reticulum; LDL – low-density lipoprotein
Diagnostika NP-C a nové laboratorní postupy
V současnosti používaná vyšetření a jejich výpovědní hodnoty jsou popsány níže.
U pacienta s klinickým podezřením na NP-C je obvykle prvním laboratorním testem vyšetření některého z biomarkerů, v případě pozitivního výsledku následuje sekvenování genů NPC1, případně i NPC2. Pokud je výsledek vyšetření biomarkerů negativní, sekvenování se provádí jen v případě velmi výrazného klinického podezření na NP-C. Nález patogenních nebo pravděpodobně patogenních mutací [15] diagnózu NP-C potvrdí a další vyšetření se již obvykle neprovádí. U variant sekvence s nejasným vlivem na fenotyp je třeba zvážit provedení dalších biochemických vyšetření, vč. funkčních testů, které mohou prokázat poruchu nitrobuněčného transportu cholesterolu. Pokud je nalezena jen jedna patogenní varianta v genech NPC1 nebo NPC2, jsou na místě další molekulárně genetická vyšetření, vč. vyšetření počtu kopií NPC1 nebo NPC2 a vyšetření transkriptu, případně i funkční test nebo vyšetření dalších biomarkerů. Souhrnná interpretace u pacientů s hraničními výsledky biochemických analýz nebo u variant s nejasným vlivem na fenotyp může být obtížná. Je proto vhodné, aby tato vyšetření provádělo specializované pracoviště, které má zkušenost s laboratorní diagnostikou NP-C. Diagnóza NP-C se považuje za potvrzenou u pacientů nesoucích dvě jasně patogenní varianty nebo majících opakovaně pozitivní výsledek funkčního testu. Velmi pravděpodobná je u pacientů s dvěma doposud nepopsanými variantami o neznámé patogenitě a hraničními výsledky funkčního testu [16].
Postup vyšetření a interpretace výsledků podrobně shrnuje nedávné expertní doporučení [16].
Biochemická diagnostika poruch intracelulárního transportu cholesterolu
Hlavním nástrojem pro definitivní laboratorní diagnostiku NP-C byl donedávna klasický filipinový test, který kvalitativně hodnotí množství volného cholesterolu zobrazeného fluorescenčním barvivem filipinem v cytoplazmě kultivovaných kožních fibroblastů (obr. 2), a to nejlépe po zátěži LDL. Test rozpoznává na základě intenzity fluorescence v cytoplazmě a procenta pozitivních buněk u NP-C tři fenotypy: klasický, intermediální a atypický variantní s menším počtem pozitivních buněk. Klasický a intermediální obraz jsou snadno rozpoznatelné a diagnostické, variantní fenotyp může být obtížnější jednoznačně vyhodnotit a může vyžadovat i porovnání s kulturami rodičů pacienta, protože mírně abnormální výsledky mohou být přítomny i u heterozygotů.
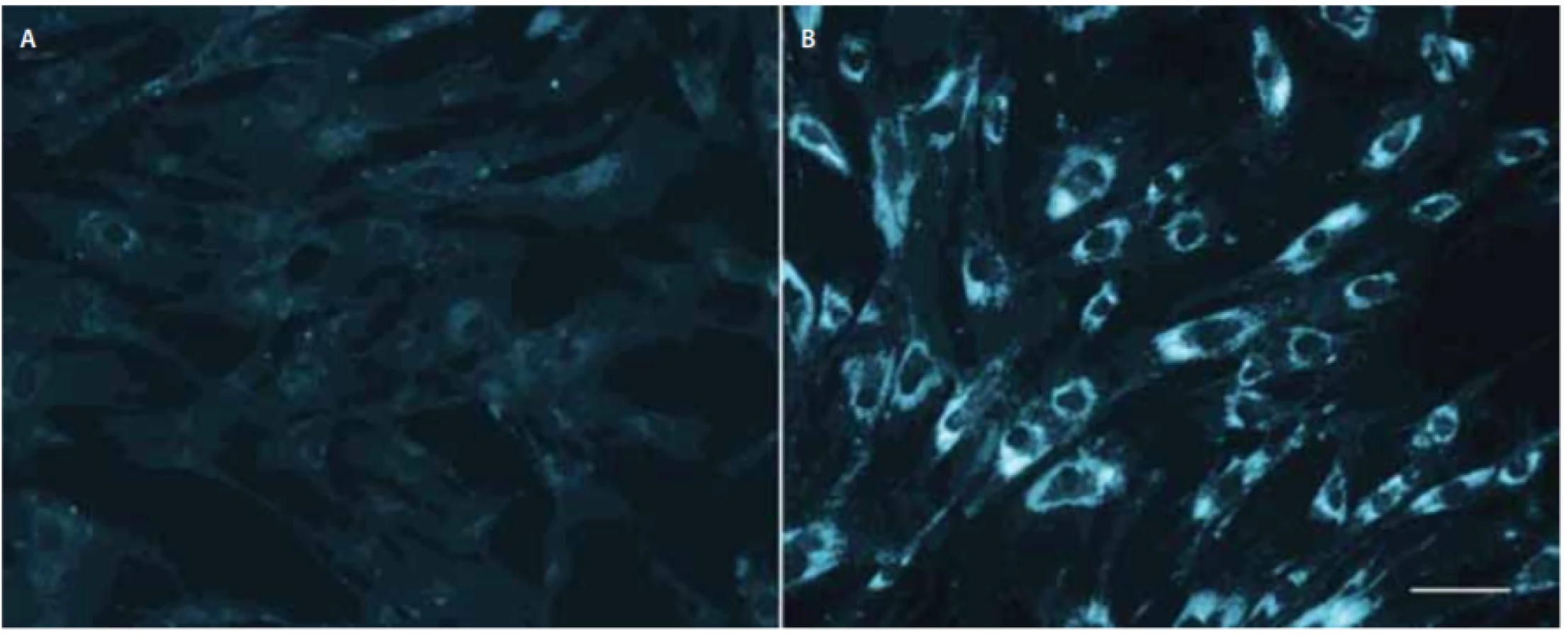
v NPC1. Délka značky v pravé části odpovídá 50 μm.
Fig. 2. Filipin staining of skin fi broblast cell cultures without low-density lipoprotein-challenge. Left panel (A) control fibroblasts; right
panel (B) fi broblasts from an infantile Niemann-Pick disease type C1 patient carrying two null mutations in NPC1. Marker on the right
corresponds to 50 μm.
Nevýhodami testu jsou pracnost, zdlouhavost, invazivní odběr kůže pro založení kultury fibroblastů a zejména expertní vyhodnocení mikroskopického obrazu. Přestože u části pacientů s „variantním“ fenotypem filipinový test nedokáže sám o sobě jednoznačně stanovit diagnózu, je považován za velmi citlivý nástroj pro diagnostiku NP-C. Falešně pozitivní výsledek byl pozorován u mukolipidózy II a slabě pozitivní nález u několika málo dalších stavů [17]. V diagnostice je nahrazován snadněji dostupnými testy s možností kvantitativního vyhodnocení. Byla popsána jeho varianta s využitím automatizované analýzy obrazu [18].
Jak je uvedeno výše, cholesterol z LE/LY je transportován také do endoplazmatického retikula, kde je esterifikován. Transport je závislý na NPC1 a NPC2, jejich deficit proto vede ke snížení esterifikace cholesterolu. Vyšetření esterifikace cholesterolu měří množství cholesterylesterů vytvořených po přidání radioaktivní kyseliny olejové a LDL. Míra snížení esterifikace koreluje s tíží klinického postižení u NP-C [19]. Varianta esterifikačního testu je vyvíjena v naší laboratoři a je založena na měření esterifikace cholesterolu značeného deuteriem, který je endocytován do buněk v tkáňové kultuře v částicích připravených v laboratoři z polysacharidu a cholesterolu. Výhodou je možnost kvantifikace výsledku a snadná a standardizovaná příprava částic pro zátěžový funkční test.
Biomarkery v tělních tekutinách – současný přehled
Aktivity chitinázy chitotriosidázy, které jsou masivně zvýšeny u Gaucherovy choroby, jsou mírně zvýšeny i v séru pacientů s NP-C. Test nemá pro NP-C dostatečnou specificitu a senzitivitu. U zhruba 30 % pacientů s NP-C je falešně negativní, téměř výhradně u pacientů s pozdními formami onemocnění (juvenilní, adolescentní/adultní). U časných forem je tento levný a dostupný test vcelku spolehlivý [20]. Nevýhodou je i klinicky němý deficit aktivity, častý v řadě evropských populací [21]. U osob s deficitem nelze výsledek hodnotit. Protože test bývá pozitivní u dalších lysozomálních onemocnění [22], je stále užitečný, např. při iniciálním vyšetření pacientů s málo specifickou klinickou symptomatologií.
Mezi nové a specifičtější biomarkery NP-C patří oxysteroly, lyzosfingolipidy a žlučové kyseliny, které lze stanovit v plazmě a někdy i v suchých krevních kapkách pomocí tandemové hmotnostní spektrometrie. Koncentrace některých oxysterolů, oxidačních produktů cholesterolu, jsou v plazmě pacientů s NP-C zvýšené. Cholestan-3b,5a,6b--triol (Triol) a 7-ketocholesterol (7-KC) jsou senzitivními a poměrně spolehlivými markery NP-C [14]. Jejich nevýhodou je možná falešná pozitivita signalizující NP-C u dalších onemocnění, zejména deficitu kyselé sfingomyelinázy a deficitu kyselé lipázy, v menší míře u cerebrotendinózní xantomatózy a syndromu Smith-Lemli-Opitz. Falešně vyšší koncentrace mohou být také přítomny u neonatální cholestázy. U těchto pacientů proto nejsou Triol a 7-KC vhodnými markery při podezření na NP-C. Zvýšení uvedených oxysterolů u deficitu kyselé sfingomyelinázy (Niemann-Pickova choroba typu A a B, NP-A/B) je podstatnou překážkou pro diferenciální diagnostiku NP-A/B a NP-C pomocí těchto markerů [23]. Další nevýhodou je relativně složitý postup přepravy a přípravy vzorků, který musí dodržovat podmínky zabraňující oxidaci cholesterolu, a tím možnému falešnému zvýšení hodnot těchto biomarkerů. Proto jsou mnohem častěji využívány biomarkery tzv. druhé generace, u kterých tato omezení nejsou.
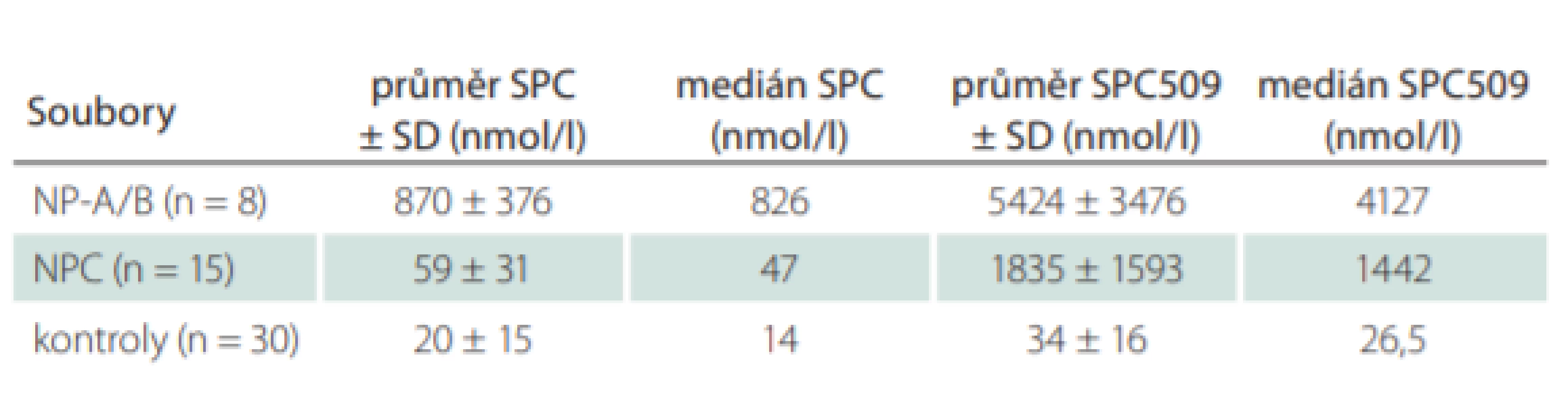
Významnými nedávno zavedenými markery jsou lyzosfingomyelin (sfingosylfosforylcholin; SPC) a tzv. lyzosfingomyelin 509 (SPC509), jehož struktura (N-acyl-O-fosfocholin serin) byla zatím objasněna pouze nepřímou strukturní analýzou [24]. Oba jsou citlivými markery, jejichž koncentrace jsou zvýšeny u NP-C a u deficitu kyselé sfingomyelinázy (NP-A/B). SPC je výrazně zvýšen téměř výhradně jen u deficitu kyselé sfingomyelinázy. Při současném stanovení obou markerů v plazmě nebo séru je tak ve většině případů možná diferenciální diagnostika NP-C a NP-A/B [25] – viz tab. 1. U sporných případů lze deficit kyselé sfingomyelinázy snadno vyloučit stanovením její aktivity. Při využití těchto biomarkerů je potřeba věnovat zvýšenou pozornost pacientům po splenektomii, protože existuje podezření, že u nich budou tato vyšetření falešně negativní [26].
Další generací markerů, které je možné analyzovat v plazmě nebo i v krevních papírcích, jsou žlučové kyseliny – 3b,5a,6b-trihydroxycholanová kyselina a glycinový konjugát kyseliny trihydroxycholanové [27,28]. Oba markery jsou mnohem specifičtější a stabilnější, protože nevznikají jako produkty arteficiální oxidace během přepravy a zpracování vzorku jako oxysteroly a nemají tak výraznou tendenci k falešně pozitivním výsledkům u neonatální cholestázy. Samotné stanovení je snadnější, protože není zapotřebí derivatizace. Tyto markery jsou velmi slibné, zatím je však jejich stanovení omezeně dostupné a nebylo plně prospektivně validováno.
Vyšetření nukleových kyselin
Sekvenování genů NPC1 a NPC2 je dobře dostupné a je někdy prvním cíleným vyšetřením při podezření na NP-C. Metodou volby je Sangerovo sekvenování. Geny NPC1 a NPC2 jsou však i součástí různých diagnostických panelů cílených na pacienty s neurologickými onemocněními a sekvenují se také při vyšetření exomu a genomu. Užitečné může být i vyšetření počtu kopií a analýza mRNA (messenger RNA). Vyšetření mutací je také jediným spolehlivým testem pro určení heterozygocie u rodinných příslušníků.
Morfologická diagnostika
Morfologická diagnostika má při vyšetřování pacientů s podezřením na NP-C velmi omezený význam díky dostupnosti biochemických a molekulárně genetických vyšetření nevyžadujících invazivní odběr vzorků tkání. Nicméně je důležité zvažovat NP-C v diferenciální diagnostice, zejména při vyšetřování nátěru kostní dřeně a při vyšetřování biopsie jater pro neonatální hepatopatii, kde v terénu cholestázy může být lysozomální střádání obtížně rozpoznatelné. V kostní dřeni jsou u pacientů s NP-C často přítomny pěnité histiocyty (diskuze v [29]), v literatuře se uvádí jako diagnosticky přínosné i barvení nátěru na filipin [30]. Histologické a elektronmikroskopické vyšetření v dalších tkáních (zejména ve slezině, v mozku a případně v lymfatických uzlinách) může v případě jejich dostupnosti napomoci stanovení diagnózy. Histologický nález v játrech je u NP-C s výjimkou výše uvedené formy neonatální cholestatické hepatopatie málo výrazný [31]. Elektronově mikroskopické vyšetření kožní biopsie, přínosné u vybraných lysozomálních střádacích onemocněních, není u NP-C diagnosticky dostatečně výpovědní.
Skríning ve vybraných skupinách pacientů
Vyšetření biomarkerů v tělesných tekutinách pomocí tandemové hmotnostní spektrometrie lze automatizovat. Stanovení oxysterolů a SPC509 v krevních papírcích není zcela spolehlivé, mimo jiné i díky částečnému překryvu hodnot u kontrol a pacientů, proto je nevhodné je pro skríning používat. Přesto je možné využít jejich stanovení v plazmě nebo séru ve skríningových programech u skupin pacientů s možným výskytem NP-C, jakými jsou např. dospělí pacienti s neuropsychiatrickými onemocněními [32]. Pozitivní skríningové nálezy je třeba ověřit dalšími technikami. V současnosti byla tato možnost využita pouze v několika studiích [33,34]. Vzhledem k běžnému mnohaletému opoždění diagnózy by včasný skríning mohl vést k dřívějšímu nasazení léčby [35].
Dostupnost vyšetření v ČR
Biomarkery – Na našem pracovišti se na základě klinického podezření na NP-C provádí stanovení biomarkerů SPC a SPC509, které mají v praxi pro efektivní diagnostiku NP-C zatím největší význam. Vyšetření je dostupné po telefonické konzultaci s lékařem Metabolického centra Kliniky dětského a dorostového lékařství 1. LF a VFN (2 2496 7710). Ročně jsou tímto způsobem vyšetřeny desítky pacientů, od listopadu 2016 do ledna 2020 bylo z cca 260 vzorků plazmy nebo séra zachyceno devět nových pacientů s NP-C a dva pacienti s NP-A/B. Stanovení nyní provádíme pouze v séru nebo plazmě a nikoli v suchých krevních kapkách, kde je nespolehlivé. Příklady výsledků u pacientů s NP-C a NP-A/B jsou uvedeny tab. 1. Je zde dostupné i vyšetření aktivity chitotriosidázy a také kyselé sfingomyelinázy, které je důležité pro vyloučení NP-A/B.
Molekulárně genetická vyšetření – Naše pracoviště rutinně provádí vyšetření genů NPC1 a NPC2 pomocí Sangerova sekvenování. Další vyšetření, např. analýza transkriptu nebo vyšetření počtu kopií, jsou možná po domluvě. Oba geny jsou součástí panelů vyšetřovaných technikami sekvenování nové generace (next generation sequencing; NGS) na řadě pracovišť v ČR, která také mohou nabízet i vyšetření exomu nebo genomu. Databázi dostupných vyšetření spravuje Společnost lékařské genetiky a genomiky [36].
Filipinový test – Tento test pro diagnostické účely žádná laboratoř v ČR neprovádí.
Závěr
Diagnostika NP-C je komplementární. Žádný z testů sám o sobě nemusí stačit pro jednoznačné stanovení diagnózy NP-C. DNA diagnostika je dostačující např. při nálezu známých patogenních mutací u pacienta s typickým klinickým obrazem, ale další výše diskutované diagnostické a laboratorní techniky mohou být zásadní u osob s minimálními nebo netypickými příznaky, které nesou v genech NPC1 nebo NPC2 varianty o neznámé patogenitě [23].
U pacientů s neurologickým onemocněním nejasného původu nebo s širokou diferenciální diagnostikou se nyní běžně jako základní vyšetření provádí vyšetřování panelů genů nebo exomu technikou NGS. Pacient, u něhož jsou nalezeny v genech NPC1 nebo NPC2 varianty o nejasné patogenitě, bývá pak doporučen k vyloučení NP-C na specializované pracoviště. Protože u takových pacientů nemusí klinická a zobrazovací vyšetření podezření vyloučit, diagnostika se opírá o metody prokazující abnormální transport cholesterolu nebo o biomarkery. Potvrzení diagnózy je rozhodující pro případné zahájení léčby u těchto pacientů. Je proto nezbytné, aby specializovaná laboratoř měla k dispozici biochemické testy, které ve sporných případech mohou diagnózu NP-C potvrdit či vyloučit.
Konflikt zájmů
Autoři deklarují, že v souvislosti s předmětem studie nemají žádný konflikt zájmů.
Grantová podpora
Podpořeno z programového projektu Ministerstva zdravotnictví ČR s reg. č. 16-33923A.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
MUDr. Martin Hřebíček
Laboratoř pro studium vzácných nemocí Klinika dětského a dorostového lékařství
1. LF UK a VFN v Praze Ke Karlovu 455
120 00 Praha
e-mail: martin.hrebicek@lf1.cuni.cz
Přijato k recenzi: 18. 2. 2020
Přijato do tisku: 29. 4. 2020
Sources
1. Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis 2010; 5: 16. doi: 10.1186/1750-1172-5-16.
2. Jahnová H, Dvořáková L, Hůlková H et al. Diagnostika a možnosti léčby Niemann-Pickovy choroby typ C. Cesk Slov Neurol N 2012; 75/108 (3): 303–308.
3. Nadjar Y, Hütter-Moncada AL, Latour P et al. Adult Niemann-Pick disease type C in France: clinical phenotypes and long-term miglustat treatment effect. Orphanet J Rare Dis 2018; 13 (1): 175. doi: 10.1186/s13023-018-0913-4.
4. Mengel E, Klünemann HH, Lourenço CM et al. Niemann-Pick disease type C symptomatology: an expert-based clinical description. Orphanet J Rare Dis 2013; 8: 166. doi: 10.1186/1750-1172-8-166.
5. Wijburg FA, Sedel F, Pineda M et al. Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology 2012; 78 (20): 1560–1567. doi: 10.1212/WNL.0b013e3182563b82.
6. Pineda M, Mengel E, Jahnová H et al. A Suspicion Index to aid screening of early-onset Niemann-Pick disease Type C (NP-C). BMC Pediatr 2016; 16: 107. doi: 10.1186/s12887-016-0641-7.
7. Patterson MC, Garver WS, Giugliani R et al. Long-term survival outcomes of patients with Niemann-Pick disease type C receiving miglustat treatment: a large retrospective observational study. J Inherit Metab Dis 2020 Apr 23 [Online ahead of print]. doi: 10.1002/jimd.12245.
8. Farmer CA, Thurm A, Farhat N et al. Long-term neuropsychological outcomes from an open-label phase I/IIa trial of 2-hydroxypropyl-b-cyclodextrins (VTS-270) in Niemann-Pick disease, type C1. CNS Drugs 2019; 33 (7): 677–683. doi: 10.1007/s40263-019-00642-2.
9. Kirkegaard T, Gray J, Priestman DA et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci Transl Med 2016; 8 (355): 355ra118. doi: 10.1126/scitranslmed.aad9823.
10. Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232 (4746): 34–47. doi: 10.1126/science.3513311.
11. Pfeffer SR. NPC intracellular cholesterol transporter 1 (NPC1) -mediated cholesterol export from lysosomes. J Biol Chem 2019; 294 (5): 1706–1709. doi: 10.1074/jbc.TM118.004165.
12. Ikonen E. Mechanisms of cellular cholesterol compartmentalization: recent insights. Curr Opin Cell Biol 2018; 53: 77–83. doi: 10.1016/j.ceb.2018.06.002.
13. Vanier MT. Complex lipid trafficking in Niemann-Pick disease type C. J Inherit Metab Dis 2015; 38 (1): 187–199. doi: 10.1007/s10545-014-9794-4.
14. Porter FD, Scherrer DE, Lanier MH et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med 2010; 2 (56): 56ra81. doi: 10.1126/scitranslmed.3001417.
15. Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17 (5): 405–423. doi: 10.1038/gim.2015.30.
16. Geberhiwot T, Moro A, Dardis A et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis 2018; 13 (1): 50. doi: 10.1186/s13023-018-0785-7.
17. Vanier MT, Latour P. Laboratory diagnosis of Niemann-Pick disease type C: the filipin staining test. Methods Cell Biol 2015; 126: 357–375. doi: 10.1016/bs.mcb.2014.10.028.
18. Tängemo C, Weber D, Theiss S et al. Niemann--Pick Type C disease: characterizing lipid levels in patients with variant lysosomal cholesterol storage. J Lipid Res 2011; 52 (4): 813–825. doi: 10.1194/jlr.P013524.
19. Vanier MT, Wenger DA, Comly ME et al. Niemann--Pick disease group C: clinical variability and diagnosis based on defective cholesterol esterification: a collaborative study on 70 patients. Clin Genet 1988; 33 (5): 331–348. doi: 10.1111/j.1399-0004.1988.tb03460.x.
20. Vanier MT, Gissen P, Bauer P et al. Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review. Mol Genet Metab 2016; 118 (4): 244–254. doi: 10.1016/j.ymgme.2016.06.004.
21. Piras I, Melis A, Ghiani ME et al. Human CHIT1 gene distribution: new data from Mediterranean and European populations. J Hum Genet 2006; 52 (2): 110–116. doi: 10.1007/s10038-006-0086-1.
22. Michelakakis H, Dimitriou E, Labadaridis I. The expanding spectrum of disorders with elevated plasma chitotriosidase activity: an update. J Inherit Metab Dis 2004; 27 (5): 705–706. doi: 10.1023/b: boli.0000043025.17721.fc.
23. Vanier MT, Gissen P, Bauer P et al. Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review. Mol Genet Metab 2016; 118 (4): 244–254. doi: 10.1016/j.ymgme.2016.06.004.
24. Sidhu R, Mondjinou Y, Qian M et al. N-acyl-O-phosphocholineserines: structures of a novel class of lipids that are biomarkers for Niemann-Pick C1 disease. J Lipid Res 2019; 60 (8): 1410–1424. doi: 10.1194/jlr.RA119000157.
25. Kuchar L, Sikora J, Gulinello ME et al. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases. Anal Biochem 2017; 525: 73–77. doi: 10.1016/j.ab.2017.02.019.
26. Lipiński P, Kuchar L, Zakharova EY et al. Chronic visceral acid sphingomyelinase deficiency (Niemann-Pick disease type B) in 16 Polish patients: long-term follow-up. Orphanet J Rare Dis 2019; 14 (1): 55. doi: 10.1186/s13023-019-1029-1.
27. Jiang X, Sidhu R, Mydock-McGrane L et al. Development of a bile acid-based newborn screen for Niemann-Pick disease type C. Sci Transl Med 2016; 8 (337): 337ra63. doi: 10.1126/scitranslmed.aaf2326.
28. Jiang X, Sidhu R, Orsini JJ et al. Diagnosis of niemann--pick C1 by measurement of bile acid biomarkers in archived newborn dried blood spots. Mol Genet Metab 2019; 126 (2): 183–187. doi: 10.1016/j.ymgme.2018.08.007.
29. Elleder M. Diagnosis of Niemann–Pick type C (NPC) – decisions at the cell level. Pathologist’s report. Mol Genet Metab 2010; 99 (1): 98. doi: 10.1016/j.ymgme.2009.09.011.
30. Wraith JE, Baumgartner MR, Bembi B et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab 2009; 98 (1–2): 152–165. doi: 10.1016/j.ymgme.2009.06.008.
31. Kelly DA, Portmann B, Mowat AP et al. Niemann-Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J Pediatr 1993; 123 (2): 242–247. doi: 10.1016/S0022-3476 (05) 81695-6.
32. Hendriksz CJ, Anheim M, Bauer P et al. The hidden Niemann-Pick type C patient: clinical niches for a rare inherited metabolic disease. Curr Med Res Opin 2017; 33 (5): 877–890. doi: 10.1080/03007995.2017.1294054.
33. Bauer P, Balding DJ, Klünemann HH et al. Ge-netic screening for Niemann-Pick disease type Cin adults with neurological and psychiatric symptoms: findings from the ZOOM study. Hum Mol Genet 2013; 22 (21): 4349–4356. doi: 10.1093/hmg/ddt284.
34. Wassif CA, Cross JL, Iben J et al. High incidence of unrecognized visceral/neurological late-onset Niemann--Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet Med 2016; 18 (1): 41–48. doi: 10.1038/gim.2015.25.
35. Sobrido MJ, Bauer P, de Koning T et al. Recommendations for patient screening in ultra-rare inherited metabolic diseases: what have we learned from Niemann--Pick disease type C? Orphanet J Rare Dis 2019; 14 (1): 20. doi: 10.1186/s13023-018-0985-1.
36. Společnost lékařské genetiky a genomiky České lékařské společnosti Jana Evangelisty Purkyně. Databáze genetických pracovišť. Dostupné z URL: https: //slg.cz/pracoviste/.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2020 Issue 3
Most read in this issue
- Glioblastom grade IV – dlouhodobé přežití
- Bolesti hlavy v graviditě
- Primární progresivní afázie
- Kognitivní poruchy u dětí s epilepsií