
Nádory očnice
Orbital Tumors
Orbital tumors constitute a relatively rare and heterogeneous group of lesions, either originating in the orbit or with secondary extension into the bony orbital margins. Primary orbital tumors are scarcer and usually benign, while secondary tumors are usually malignant. Metastases may be found anywhere in the wall of the orbit or inside the orbit. The most common symptoms include exophtalmus, visual impairment and diplopia. CT in combination with MRI are the most useful in the diagnostic algorithm. Therapy aims for long-term management of tumor growth with focus on visual improvement or preservation. Monitoring, surgery, chemotherapy and radiation with respect to histology and staging are all used in combination as it is common in other groups of tumors. Over the past few decades, advances in radiodiagnostics, improved knowledge of the natural course of tumors, more accurate radiosurgical techniques and improved chemotherapy protocols have changed therapeutic algorithms as well as prognosis in many orbital tambours. Multidisciplinary approach in specialized centers shall ensure optimal treatment.
Key words:
orbit – orbital tumor – exophtalmus – orbitotomy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
:
P. Vachata 1,2; L. Zikmund 3; J. Kozák 1; F. Cihlář 4; D. Šulc 5; M. Sameš 1,2
:
Neurochirurgická klinika UJEP a Krajská zdravotní a. s., Masarykova nemocnice v Ústí nad Labem, o. z.
1; Skull base center Class A, World Federation of Neurosurgical Societies, Ústí nad Labem
2; Oční klinika UJEP a Krajská zdravotní a. s., Masarykova nemocnice v Ústí nad Labem, o. z.
3; Radiodiagnostické oddělení, Krajská zdravotní a. s., Masarykova nemocnice v Ústí nad Labem, o. z.
4; Komplexní onkologické centrum, Radioterapeutické oddělení, Krajská zdravotní a. s., Masarykova nemocnice v Ústí nad Labem, o. z.
5
:
Cesk Slov Neurol N 2015; 78/111(6): 617-638
:
Minimonography
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amcsnn2015617
Nádory očnice tvoří relativně vzácnou a velmi heterogenní skupinu nádorových expanzí primárně uložených nebo sekundárně se propagujících do prostoru ohraničeného kostěnou orbitou. Primární orbitální nádory jsou vzácnější a většinou benigní, zatímco sekundární nádory mají spíše maligní charakter. Metastázy mohou být uloženy kdekoliv ve stěně anebo uvnitř orbity. Klinickými projevy tumorů jsou nejčastěji exoftalmus, poruchy zraku a diplopie. Nejpřínosnější vyšetření orbitálních patologií je kombinace CT a MR. Cílem léčby je dlouhodobá kontrola nádoru se snahou o uchování nebo zlepšení zraku. V terapii se využívá stejně jako u jiných nádorů observace, chirurgie, chemoterapie i radioterapie s ohledem na histologickou specifikaci a staging. Pokroky v radiodiagnostice, vzrůstající znalosti o přirozeném průběhu onemocnění, přesné radiochirurgické techniky a moderní chemoterapeutické protokoly změnily v posledních desetiletích terapeutické postupy i prognózu u celé řady nádorů očnice. Optimální je léčba vedená prostřednictvím multidisciplinárních týmů ve specializovaných centrech zabývajících se touto problematikou.
Klíčová slova:
orbita – nádor orbity – exoftalmus – orbitotomie
Úvod
Orbitální tumory tvoří relativně vzácnou a velmi heterogenní skupinu nádorových expanzí uložených nebo propagujících se do prostoru ohraničeného kostěnou očnicí. Vzhledem k tomu, že vlastní očnice je integrální součástí spodiny lebeční s těsnými anatomickými vztahy jak k nitrolebnímu prostoru, tak i k ostatním přilehlým extrakraniálním kompartmentům, je chirurgická problematika těchto lézí typický příklad multidisciplinární chirurgie lební báze. Stejně tak jako je rozmanitá paleta těchto patologií, je rozmanité i spektrum odborností zabývajících se jejich léčbou. Složitá anatomie očnice a komplexní topografické vztahy kladou významné nároky nejen na diferenciální diagnostiku a hodnocení grafického zobrazení, ale především na vlastní terapeutický management realizovaný ideálně prostřednictvím multidisciplinárního týmu. Kromě neurochirurgů jsou tyto nádory řešeny otorinolaryngology, stomatochirurgy a oftalmology. Doménou oftalmologů jsou vlastní nádory oční koule (melanom, retinoblastom), které tvoří oddělenou skupinu patologií, jež není předmětem tohoto sdělení, ačkoli bývá někdy řazena mezi sekundární nádory očnice [1]. Vzhledem ke klinické symptomatologii bývá oftalmolog nejčastěji první odborný kontakt pacienta. Mezi další neoddělitelné členy multidisciplinárního týmu patří radiologové a onkologové. Separátní skupina nádorových lézí v této oblasti jsou nádory kožní v oblasti očních víček a bezprostředním okolí očnice. Tuto onkologickou problematiku řeší plastičtí chirurgové a oftalmologové specializující se na okuloplastickou chirurgii. Vzhledem k heterogenitě těchto lézí i odborností zabývajících se jejich léčbou je obtížné stanovit jejich přesný výskyt v populaci. Při analýze vykazovaných výkonů celkem 19 neurochirurgických pracovišť je udáváno každý rok pouze mezi 20 a 25 operovanými případy nádorů očnice v celé České republice. Fakt, že tyto výkony tvoří méně než promile všech operačních neurochirurgických výkonů, potvrzuje vzácnost těchto patologií. Autoři v této monografii vycházejí z vlastního souboru 72 pacientů operovaných pro expanzi očnice v letech 1999–2014 na Neurochirurgické klinice v Ústí nad Labem.
Historicky prvním sofistikovaným chirurgickým přístupem do orbity byla prostá přední orbitotomie popsaná Knappem v roce 1874 [2]. Laterální orbitotomie publikovaná Krönleinem v roce 1889 umožnila atakovat intrakonální léze [3]. Teprve s významným odstupem v roce 1941 W. E. Dandy, jeden z pionýrů neurochirurgie, hodnotil řešení intraorbitálních patologií transkraniálním přístupem, který poprvé použil v roce 1921 [4]. Jeho přístup se zachováním hrany orbity vycházel z Frazierova transkraniálního přístupu k hypofýze využívajícího orbitální osteotomii [5]. Kromě zavedení mikrochirurgických technik v 60. letech minulého století bylo dalším významným mezníkem v chirurgii očnice použití endoskopických technik Norrisem a Cleasbym v roce 1981 [6]. V radiodiagnostice byla zásadním přelomem po zavedení CT především implementace MR vyšetření k detailnímu posouzení měkkých tkání orbity [7]. Kvalitní radiodiagnostika, vzrůstající znalosti o přirozeném průběhu onemocnění, přesné stereoradiochirurgické techniky a moderní chemoterapeutické protokoly dokázaly v posledních desetiletích změnit terapeutické postupy u celé řady patologií očnice. Příkladem může být preference chemoterapie v terapii gliomů optiku, preference radioterapie u meningeomů pochvy optického nervu nebo odklon od mutilujících exenterací u malignit vedlejších dutin nosních propagujících se do očnice [7–13]. V našem domácím písemnictví nelze opominout dosud jedinou komplexní monografii Otradovce z roku 1986 zabývající se chorobami očnice vč. orbitálních nádorů [14].
Anatomie orbity
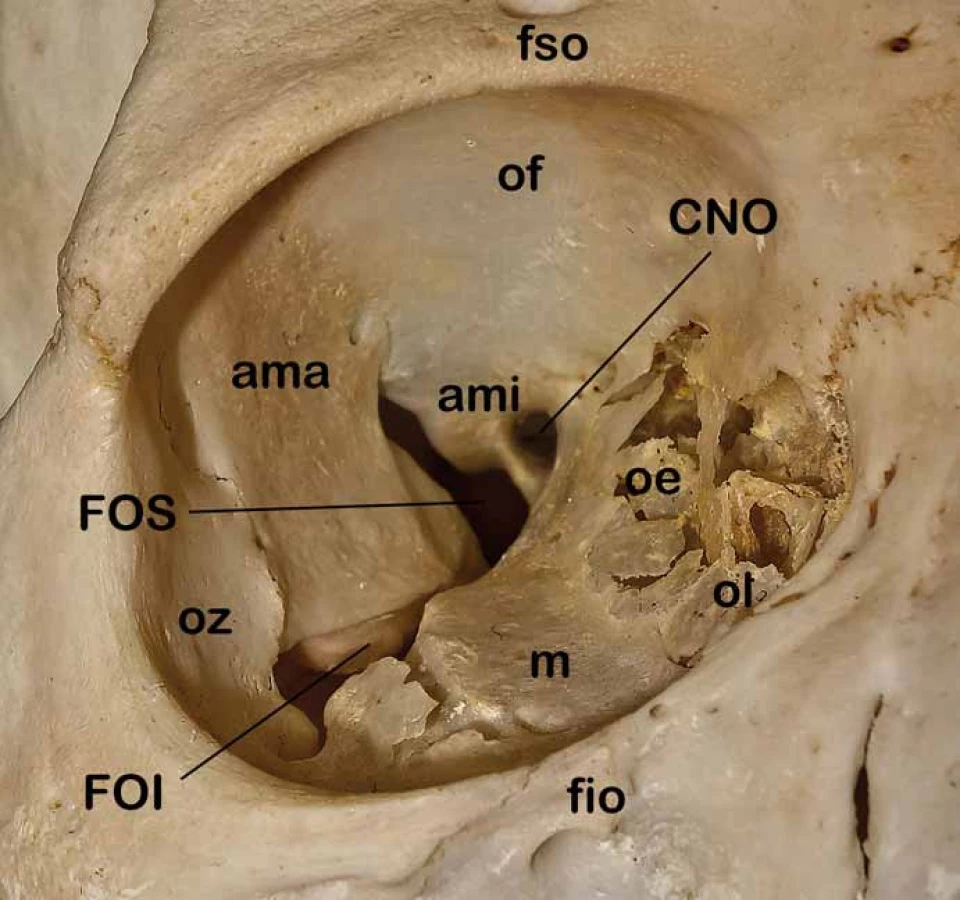
Znalost topografické anatomie je základní předpoklad managementu nádorů očnice [15–17]. Orbita obsahuje oční bulbus, okohybné svaly a oční adnexa. Kostěná očnice vymezující intraorbitální prostor je tvořena několika kostěnými strukturami (os frontale, os zygomaticum, maxilla, os lacrimale, os sphenoidale, os palatinum, os ethmoidale) spojenými odpovídajícími kostěnými švy. Očnice má typický konický tvar čtyřboké pyramidy s apexem směřujícím kraniálně do oblasti optického kanálu a se zevním okrajem neboli vchodem (aditus orbitae) směřujícím anteriorně s mírnou laterální a inferiorní inklinací. Orbitální vchod tvořený orbitální hranou (margo orbitalis) má průměrnou šířku 41 mm a výšku 34 mm. Nejširší místo očnice je však uloženo asi centimetr za vlastním vchodem. Hloubka očnice je kolem 42 mm. Pro mediální stěny, které jsou od sebe v průměrné intraorbitální vzdálenosti kolem 25 mm, je typické paralelní postavení na rozdíl od laterálních stěn, které divergují (obr. 1). Mediální stěna je tvořena frontálním výběžkem maxilly, slznou kostí se svislou brázdou slzného vaku kaudálně přecházející v nazolakrimální vývod, kostí čichovou a malým křídlem kosti klínové. Nejtenčí je mediální stěna v místě sousedícím s čichovými sklípky (lamina papyracea ossis ethmoidalis), která může být i kongenitálně dehiscentní. V mediální stěně orbity můžeme identifikovat foramen ethmoidale anterius et posterius obsahující stejnojmenné tepny a větve nazociliárního nervu. Strop očnice je tvořen obdobně tenkou orbitální částí frontální kosti a vzadu částí malých křídel kosti klínové. Mediálně je strop spodinou čelní dutiny, která může být často zdvojena supraorbitálním recesem, vzadu je pak strop spodinou přední jámy lební. Laterálně vpředu lze identifikovat prohlubeň pro slznou žlázu (fossa glandulae lacrimalis) a naopak mediálně vpředu fossa trochlearis, kde je fixována chrupavčitá kladka pro horní šikmý sval. Na rozhraní vnitřní a střední třetiny je v orbitálním okraji supraorbitální incisura obsahující stejnojmenný nervově cévní svazek. Spodina očnice je tvořena z větší části tenkou kostí horní čelisti (facies orbitalis maxillae), dále okrajem lícní kosti a orbitálním výběžkem kosti patrové. Spodní stěna je hladká a obsahuje infraorbitální žlábek, který se prohlubuje v infraorbitální kanálek s nervově-cévním infraorbitálním svazkem. Spodinu z větší části tvoří strop maxillární dutiny. Laterální stěna je ze všech stěn nejtlustší a je tvořena vpředu lícní kostí a vzadu velkým křídlem kosti klínové. V přední třetině sousedí s temporální jámou a v zadních dvou třetinách se střední jámou lební. Ve vrcholu očnice je uložen v malém křídle kosti klínové optický kanálek oválného průřezu o velikosti 5 × 4,5 mm a délce 8–10 mm (obr. 2). Kanálek je ve sfenoidální dutině lokalizován ve stěně v úhlu mezi stropem a laterální stěnou a intrakraniálně vstupuje inferomediálně od processus clinoideus anterior. Kanálkem prochází nervus opticus spolu s oftalmickou tepnou, která je uložena inferomediálně pod vlastním nervem. Laterálně od optického kanálku mezi malým a velkým křídlem kosti klínové je lokalizována fissura orbitalis superior (cerebralis) spojující očnici se střední lební jámou v oblasti kavernózního splavu (obr. 2). V této štěrbině probíhají všechny okohybné nervy (horní a dolní větev n. oculomotorius, n. trochlearis a n. abducens), n. ophtalmicus (první větev n. trigeminus) ve formě svých větví (n. lacrimalis, n. frontalis, a n. nasociliaris), vlákna sympatiku, arteriální větvička a. meningea media a v. orbitalis superior komunikující s vlastním kavernózním splavem. Inferiorně od horní fissury pod velkým křídlem kosti klínové a nad maxilou se nachází fissura orbitalis inferior (sphenomaxillaris) spojující očnici s fossa pterygopalatina a fossa infratemporalis (obr. 2). Cestou této štěrbiny vstupují do očnice z fossa pterygopalatina n. zygomaticus a n. infraorbitalis dvě větve n. maxillaris (druhá větev n. trigeminus), který vstupuje do této oblasti cestou foramen rotundum. Kromě těchto nervů zde prochází také cévy a. et v. infraorbitales a v. orbitalis inferior. Kostěné stěny orbity jsou kryty periostální membránou periorbitou, která posteriorně přechází v tvrdou plenu obalující optický nerv a anteriorně v septum orbitale, jež zasahuje do očních víček a spolu s vlastním očním bulbem uzavírá očnici zevně. V oblasti obou koutků je septum fixováno ke kostěné orbitě. Kromě dalších fixací v oblasti apexu, trochley a obou etmoidálních foramin je periorbita velmi dobře separovatelná od kosti a tvoří významnou bariéru pro sekundární primárně extraorbitální expanze. Posteriorně kolem střední části fissura orbitalis superior a optického kanálku se periorbita ztlušťuje a formuje anulus tendineus communis Zinni. K tomuto vazivovému prstenci se upínají všechny čtyři přímé okohybné svaly (m. rectus lateralis, medialis, inferior a superior), m. obliquus superior a m. levator palpebrae superior. Všechny přímé svaly jsou laterálně mezi sebou spojeny mezisvalovým septem vymezujícím centrální tzv. intrakonální prostor orbity. Kromě těchto svalů je zde ještě m. obliquus inferior začínající v oblasti spodiny orbity na crista lacrimalis posterior a upínající se na bulbus v dolním laterálním zadním kvadrantu. Jeho funkcí je pohyb bulbu nahoru a laterálně. M. obliquus superior běží z oblasti společného vazivového prstence, obkružuje trochleu a upíná se na zadní laterální horní kvadrant bulbu, kterým pohybuje naopak dolů a laterálně. M. rectus medialis pohybuje bulbem mediálně a m. rectus lateralis naopak laterálně. M. rectus superior otáčí bulbus superomediálně a naopak m. rectus inferior inferomediálně. M. levator palpebrae superioris běží z anulus tendineus pod stropem orbity a končí v tarzální ploténce horního víčka, které elevuje. Kromě m. rectus lateralis inervovaného n. abducens a m. obliquus superior inervovaném n. trochlearis jsou všechny ostatní okohybné svaly inervovány n. oculomotorius. Poruchy jednotlivých svalů anebo jejich inervace se projeví odpovídající poruchou hybnosti doprovázenou diplopií. Slzný aparát je tvořen slznou žlázou a odvodnými slznými cestami. Glandula lacrimalis je uložena ve fossa glandulae lacrimalis při horním zevním obvodu stropu očnice a je rozdělena šlachou m. levator palpebrae superioris na kompaktní orbitální a nekompaktní z více drobných žlázek složenou palpebrální část. Odvodné slzné cesty začínají od punta lacrimalia v oblasti obou víček blízko vnitřního koutku jako canaliculi lacrimales superior et inferior. Oba kanálky zpravidla končí společnou ampulou v slzném vaku uloženém ve fossa sacci lacrimalis, který přechází v ductus nasolacrimalis, který ústí pod dolní skořepou nosní do dolního nosního průduchu. Dominantní arteriální zásobení očnice je cestou a. ophtalmica, která se větví na a. centralis retinae, a. lacrimalis, rr. musculares, rr. ciliares, r. supraorbitalis a aa. ethmoidales anterior et posterior. Orbitálními fissurami vstupují větve z povodí a. carotis externa a. meningea reccurens a a. infraorbitalis. V případě chronického uzávěru vnitřní karotidy se v oblasti rozvodí mezi vnitřní a vnější karotidou v oblasti orbity vytvářejí funkčně významné arteriální kolaterály zásobující cestou a. ophtalmica intrakraniální hypoperfundované povodí a. carotis interna. Dominantní žilní drenáž očnice je horní a dolní orbitální žíla. Všechen prostor očnice mezi jednotlivými strukturami a periorbitou je vyplněn orbitálním tukem, který typicky při otevření periobity defektem hřezne.

Klinické příznaky tumorů očnice
Kromě stále častějších incidentálních záchytů nádorů způsobených vyšší dostupností radiologických vyšetřovacích metod se většina nádorů očnice klinicky projeví nejčastěji rozvojem exoftalmu, poruchou zraku a diplopií [18–21]. Axiální exoftalmus je spojen s lézemi intrakonálními a v apexu orbity, zatímco nonaxiální exoftalmus spojený se stranovou dislokací bulbu svědčí pro excentrickou extrakonální lézi. Porucha zraku se zhoršením zrakové ostrosti, výpady v perimetru, ale i poruchou barevného vidění mohou být způsobeny jak přímou kompresí optického nervu, tak poškozením jeho cévního zásobení. Časná porucha zraku bez rozvoje exoftalmu budí podezření na intrakanalikulární lézi. Při vyšetření očního pozadí je možné identifikovat jak papiledém, tak atrofii optiku. Někdy je možné také zachytit patologickou vaskularizaci. Chemóza spojivek souvisí s poruchou žilní drenáže. Diplopie může být způsobena jak poruchou okohybných svalů a jejich inervace, tak mechanickým omezením hybnosti bulbu vlastní masou nádoru. Při poškození odpovídajících reflexních oblouků můžeme identifikovat poruchy přímé a nepřímé fotoreakce. Dalším častým příznakem u nádorů očnice je přítomnost bolesti, ať typicky lokalizované v oblasti orbity nebo na celé ipsilaterální straně hlavy. Při progresi nádorů nebo při primárním vzniku nádorů mimo očnici se přidávají příznaky odpovídající lokalizaci patologie (epilepsie, organický psychosyndrom, hypofyzární syndromy, diencefalický syndrom, porucha průchodnosti nosní dutiny, epistaxe). Rozvoj klinických příznaků může být akutní u zánětlivých a maligních lézí nebo pomalu progredující u lézí benigních.
Radiodiagnostika tumorů orbity
Spolu s anamnézou a oftalmologickým vyšetřením má radiodiagnostika zcela zásadní postavení v diferenciální diagnostice expanzivních procesů orbity. Speciální nativní rentgenové projekce (šikmé projekce na optický kanál dle Rheseho nebo Gravese) i negativní a pozitivní orbitografie byly již v diagnostickém schématu opuštěny [14]. Stále však je možné na prostém nativním rentgenu lbi, obličeje nebo vedlejších dutin nosních zachytit patologii s destrukcí skeletu očnice, hyperostózou, anebo zastřením fyziologicky pneumatizovaných přilehlých dutin, která by měla vést v diferenciální diagnostice k vyloučení orbitální expanze. Ultrazvuk, ačkoliv byl v diagnostice nádorů očnice vytlačen moderními zobrazovacími metodami, stále zůstává pro svoji dostupnost a nezátěžovost cennou skríningovou a monitorovací pomůckou u některých patologií, jako např. u Gravesovy endokrinní orbitopatie [22]. Bezpečně odliší cystické a solidní léze, stejně tak jako posoudí vaskularizaci v dopplerovském módu. Přínosný je také u emergentních vyšetření. Limitací jsou v apexu uložené léze a významná dependence na zkušenosti vyšetřujícího. V současné době je základ radiodiagnostiky nádorů očnice vyšetření pomocí CT a MR [23]. První zmínka o využití CT v diagnostice nádorů orbity je z roku 1977 [24]. CT v klasickém kostním okně, event. ve variantě s vysokým rozlišením (High Resolution CT; HRCT), přináší nejlepší informaci o skeletu orbity. Morfologie kostního defektu napoví o benigním nebo maligním charakteru léze. Příkladem může být hladká ostrá imprese v místě fossa lacrimalis svědčící o benigní lézi slzné žlázy na rozdíl od nepravidelné osteolytické destrukce u lézí maligních [20]. Nepostradatelná je jeho role u sekundárních tumorů orbity pro jejich přesný staging a stanovení terapeutického postupu [25]. CT bezpečně také detekuje nádorové kalcifikace a rozsah intraoseálních nádorů (meningeom, fibrózní dysplazie). Diagnostickou metodou volby pro vyšetření intraorbitálních struktur zůstává i dnes CT s aplikací jodového kontrastu u pacientů s kontraindikací MR. MR přináší detailní informaci o měkkých tkáních očnice a o vztahu patologie ke kritickým strukturám, jako je optický nerv, okohybné svaly nebo vlastní bulbus. Výhodou je na rozdíl od CT radiační nezátěžovost pro oční čočku. Kromě standardních vyšetřovacích sekvencí se v oblasti orbity využívají sekvence FLAIR spolu se sekvencemi bez potlačení signálu tuku i s ním (nativních i po podání gadoliniového kontrastu). Dalším přínosem v zobrazení patologií očnice je MR s vysokým rozlišením (high-resolution MRI) s využitím povrchových orbitálních cívek [26,27]. Kromě těchto sekvencí je možné využít řadu dalších sekvencí vylepšujících kvalitu zobrazení, jako jsou např. 3D-CISs, 3D-PSIF a 3D-VIBE sekvence [28,29]. Publikované studie na experimentálních MR naznačují, že lze v dohledné době očekávat další pokrok v zobrazení [30]. Angiografické vyšetření při dostupnosti CT a MR angiografie je dnes rezervováno pro diagnostiku cévních patologií a endovaskulární intervence (embolizace) [31]. K vyšetření metabolické aktivity lézí a posouzení stagingu se využívá pozitronová emisní tomografie [32].
Terapie nádorů orbity
Cílem multidisciplinární léčby je dlouhodobá kontrola onkologického problému při eliminaci deteriorace zrakových funkcí, nebo dokonce se snahou o jejich zlepšení při jejich vstupním poškození. Stejně jako u jakýchkoli jiných nádorů můžeme využít čtyři základní modality: observaci, chirurgii, radioterapeutické techniky a chemoterapii.
Observace je indikována obecně u benigních, pomalu rostoucích nebo dokonce stagnujících patologií bez poruchy zrakových funkcí nebo s jejich mírnou dlouhodobě stabilní deteriorací. Podmínkou observace jsou pravidelné grafické a klinické kontroly [33–35].
Indikace chemoterapie závisí především na histologickém typu patologie a její senzitivitě. Výjimkou v indikaci chemoterapie je její použití u gliomů optické dráhy, kdy k jejímu nasazení dnes zpravidla stačí typický radiologický a klinický obraz [36]. Naopak u ostatních nádorů, jako např. u lymfomů, je histologické vyšetření pro indikaci daného chemoterapeutického protokolu zcela zásadní [37].
Pro použití radioterapeutických metod je důležitá minimalizace rizika vzniku optické neuropatie s doprovázející deteriorací zraku. Na základě hodnocení výsledků radioterapeutických metod na optickou dráhu byly stanoveny prahy toxicity pro optický nerv. Leber et al stanovili incidenci vzniku neuropatie 0 %, 26,7 % a 77,8 % u optických nervů, jež obdržely dávky při radiochirurgickém ošetření gama nožem < 10 Gy, 10–15 Gy a > 15 Gy [38]. Tishler et al jako bezpečnou hranici tolerance deklarovali 8 Gy [39]. Stafford et al prokázali výskyt optické neuropatie ve skupině pod 8 Gy a 8–10 Gy shodně po 1,7 % [40]. Obdobně Mayo et al identifikovali výrazně vzrůstající riziko toxicity při dávkách převyšujících 60 Gy při 1,8 Gy na frakci a při dávce vyšší než 12 Gy při jednorázovém radiochirurgickém výkonu [41]. V současné době se proto většinou preferují u zrak šetřících výkonů frakcionované protokoly formou stereotaktické frakcionované radiochirurgie, intenzitou modulované radioterapie, stereotaktické frakcionované radioterapie nebo 3D konformní radioterapie. Díky těmto moderním metodám s lepší fokusací cílové dávky a minimalizací ozáření okolních struktur došlo k významné redukci poradiační morbidity [8,12,42–51]. Vždy je však třeba pacienta jak na přechodné akutní nežádoucí účinky (erytém, otok, alopecie, cefalea, nausea), tak na pozdní perzistující (retinopatie, optická neuropatie, katarakta, poškození cév, pituitární dysfunkce) upozornit [42].
Obecný cíl onkochirurgie je radikální resekce nádoru. Radikalita chirurgické terapie tumorů očnice a s ní spojená pooperační morbidita při snaze o zachování zrakových funkcíjelimitována postižením obalu optiku, horní orbitální fissury, orbitálního apexu s anulus tendineus a kavernózního splavu. Nutno si uvědomit, že vzniklá permanentní diplopie je pro pacienta zpravidla větší limitace než těžká monokulární porucha zrakové ostrosti. Volba chirurgického přístupu do očnice vychází z detailní předoperační analýzy topografiých vztahů nádoru, jeho velikosti, lokalizaci, charakteru, předpokládané histologie a také plánovanému cíli operačního výkonu (biopsie, parciální resekce, radikální resekce) [52–55]. Nejjednodušší výkon je biopsie k získání materiálu pro histologické vyšetření. Biopsii je možno provádět tenkou jehlou, endoskopicky nebo z limitovaného přístupu s excizí větší části patologické tkáně perkutánně či transkonjunktiválně [1,25]. K přesné identifikaci cílové tkáně se využívá navigace ultrazvukem, CT, stereotaxí nebo navigačními systémy. Historicky byla publikována i bizarní transcerebrální biopsie [56]. Pleomorfní adenom slzné žlázy a hypervaskularizované léze jsou kontraindikací biopsie [1]. Vlastní chirurgické přístupy do očnice lze rozdělit na přímé transorbitální a na přístupy extraorbitální, kdy současně pronikáme do dalšího kompartmentu, typicky intrakraniálně [18,53–55]. Orbitotomie je obecně definována jako jakýkoliv chirurgický přístup do očnice. Nejjednodušší přístup je přední orbitotomie, kdy pronikáme do očnice transpalpebrálně nebo transkonjunktiválně dominantně k lézím uloženým v přední části orbity a peribulbárně v přední polovině očnice typicky bez osteotomie [1,25]. Kožní řez při transpalpebrálních přístupech může být lokalizován v obočí, na horní hraně orbity, v záhybu horního víčka, subciliárně, v dolním záhybu víčka nebo při dolní hraně očnice. Výhodou transkonjunktiválních přístupů je absence viditelné jizvy. Laterální osteoplastická orbitotomie umožňující atakovat laterální retrobulbární extrakonální a intrakonální léze vychází z Berkeho modifikace Krönleinova původního přístupu většinou dnes s využitím esovitého kožního řezu bez laterální kantotomie (obr. 3) [1,3,52,57]. Inferiorní retrobulbárně lokalizované léze lze atakovat transantrálně cestou maxillárního sinu posteroinferiorní orbitotomií, nebo LeForte I orbitotomií [25,53]. Mediálně uložené léze v přední polovině orbity se dají atakovat mediální transkonjuntivální orbitotomií prezentovanou Galbraithem a Sullivanem v rce 1973 s peritomií a dočasným odpojením úponu mediálního přímého svalu [1,52,58]. Je možné ji kombinovat s laterální orbitotomií k získání prostoru pro mobilizaci bulbu laterálně formou tzv. mediální laterální orbitotomie [52]. Další možnost přístupu k posteriorně mediálně uloženým nádorům je získání trajektorie přístupu pomocí přední a zadní etmoidektomie [14,25]. Právě retrobulbární léze lokalizované inferiorně a mediálně od optického nervu jsou dnes atakovány endoskopickým transnazálním přístupem [59–62]. Endoskopické výkony nahradily také náročný přístup kontralaterální pterionální kraniotomií k posteriorním intrakonálním lézím uloženým mediálně a inferiorně od optického nervu [63]. Z extraorbitálních přístupů jsou nejčastěji používány některé z variant klasické frontotemporální pterionální kraniotomie (např. orbitopterionální přístup), které umožňují atakovat posteriorní laterálně a superiorně lokalizované intra - i extrakonální patologie vč. apexu, oblasti horní orbitální štěrbiny, optického kanálku a také přilehlý intrakraniální prostor (obr. 3) [1,18,53,62]. Rozšířením o zygomatickou osteotomii pak umožňují tyto přístupy atakovat i inferiorně uložené patologie [62]. Další extraorbitální přístup je jednostranný Dandyho frontoorbitální přístup [4,14,18,53]. Jeho oboustranná varianta je subfrontální přístup využívaný především u sekundárních tumorů očnice šířících se z přilehlých paranazálních dutin [25,64]. V některých případech je nutné provést výkony typu eviscerace, enukleace, exenterace nebo orbitektomie z důvodů vitální indikace, uchování zraku na druhém oku, terapie orbitální bolesti nebo korekci esteticky neakceptovatelného vzhledu [1]. Všechny tyto výkony vyžadují psychologickou podporu pacienta. Zatímco evisceraci (odstranění intraokulárního obsahu s uchováním skléry) u nádorů očnice nevyužíváme, enukleace (odstranění bulbu a intraorbitální části optického nervu) může být indikována u bolestivého slepého oka, např. u pozdních stadií gliomu optiku nebo meningeomu pochvy optického nervu (obr. 4) [1]. Typický výkon u malignit orbity je exenterace, tedy radikální odstranění veškerého orbitálního obsahu a očních víček. Při subtotální exenteraci jsou oční víčka zachována [65,66]. Rozšířenou exenteraci o resekci kostěné orbity označujeme jako orbitektomii (obr. 5) [14]. Nejradikálnější výkony jsou „en bloc“ orbitektomie, kdy celý blok orbity je odstraňován v celku bez narušení hranic nádoru [67]. Po těchto mutilujících výkonech by se mělo vždy u pacientů s předpokládanou lepší celkovou prognózou uvažovat o zhotovení protézy nebo epitézy zmírňující vzniklý kosmetický defekt. Oční protézy nahrazují pouze oční bulbus a jsou vyráběny nejčastěji ze skla nebo akrylátu. Epitézy (obličejové ektoprotézy) nahrazují kromě oční koule i chybějící periorbitální měkké tkáně. Epitézy se dnes nejčastěji vyrábějí ze silikonu nebo akrylátu a v místě defektu bývají fixovány magnety, lepidlem, kovovými čepy či pomocí brýlových obrouček [68,69].



Z technik, které zlepšují výsledky chirurgické terapie, je třeba se zmínit o použití intraoperační navigace (bezrámové neuronavigační systémy, sonografie, intraoperační zobrazovací metody) a také intraoperačního elektrofyziologického monitoringu okohybných nervů (obr. 6) [70–72]. Zatímco neuronavigační systémy jsou přínosné pro posouzení rozsahu resekce kostěné očnice, tak v případě intraorbitální navigace v nepřehledném tuku preferujeme ultrazvuk, jenž není limitován posunem struktur při otevření periorbity. Nezbytnou součástí operačního výkonu je i závěrečná rekonstrukce orbity. Periorbitu neuzavíráme vodotěsně, ale snažíme se zamezit prolapsu orbitálního tuku s rizikem vzniku pooperačního enoftalmu pomocí např. periostálních záplat. Zatímco orbitální vchod rekonstruujeme vždy pečlivě, tak v případě rekonstrukce defektů jednotlivých stěn nahrazujeme jen rozsáhlejší defekty. Se vznikem pooperačního pulzujícího exoftalmu jsme se nikdy nesetkali, neboť pro jeho vznik je nutná kromě defektu skeletu také přítomnost herniace intrakraniálního obsahu do prostoru očnice, tak jako je tomu např. po traumatech spojených s defektem tvrdé pleny nebo typicky u pacientů s neurofibromatózou typu 1 při dysplazii křídla kosti klínové [73].

Klasifikace orbitálních tumorů
Orbitální tumory můžeme dělit podle lokalizace a podle původu [18,20,21,74]. Podle lokalizace rozlišujeme tumory intrakonální lokalizované v centrálním prostoru tvořeném čtyřmi přímými okohybnými svaly a jejich intermuskulárními septy. Tumory uložené zevně svalového konu označujeme jako extrakonální anebo periferní. Nádory okupující oba kompartmenty nazýváme difuzní. Polohu expanze popisujeme dále pomocí čtyř kvadrantů (horní zevní, horní vnitřní, dolní zevní a dolní vnitřní). Jednotlivé kvadranty ještě rozdělujeme na přední a zadní podle vztahu k očnímu bulbu. Tumory posteriorní části zadních kvadrantů označujeme jako nádory orbitálního hrotu (apexu). Nádory vznikající ze struktur uložených v očnici nazýváme primární intraorbitální tumory. Tyto nádory jsou všeobecně vzácné a většinou benigní. Nádory primárně vznikající ze struktur mimo vlastní očnici a propagující se svým růstem následně do jejího prostoru označujeme za sekundární orbitální nebo také primárně extraorbitální tumory. Tato skupina nádorů je oproti primárním častější, a pokud vznikají v oblasti vedlejších dutin nosních zpravidla maligního charakteru. Zvláštní skupinu tvoří metastázy vzdálených malignit, které mohou být uloženy kdekoliv ve stěně anebo uvnitř orbity. Od skutečných nádorových lézí je nutno odlišit léze cévní a ostatní expanzivní léze označované obecně jako pseudotumory nebo také nádory imitující expanze.
Primární orbitální tumory
Gliom zrakového nervu
Gliomy optiku jsou nejčastější primární tumory optického nervu a tvoří asi 2–3 % orbitálních tumorů a 1 % všech tumorů mozku [75–77]. Vzhledem k možné topické lokalizaci je používáno souhrnné označení gliomy optické dráhy, kam zahrnujeme také gliomy optického traktu a hypothalamické gliomy. V naprosté většině se jedná histologicky o juvenilní pilocytické astrocytomy WHO grade I. Vzácně jsou popisovány fibrilární a pilomyxoidní astrocytomy, oligodendrogliomy nebo gangliogliomy [78]. Velmi vzácné maligní formy se většinou vyskytují v dospělém věku a jsou klasifikovány jako anaplastický astrocytom nebo multiformní glioblastom, přičemž se může jednat o maligní transformaci původně nízkostupňového nádoru po radioterapii [20,79]. Nejčastější výskyt je v dětské populaci mezi druhým a šestým rokem a 90 % všech tumorů se prezentuje u mladších dospělých do konce druhé dekády věku [80]. Děvčata jsou postižena mírně častěji než chlapci v poměru asi 3 : 2 [81]. Nádor asi v 50 % může být součástí autozomálně dominantní neurofibromatózy typu I (NF I). U pacientů s NF I se gliomy optiku vyskytují asi u 20–30 % pacientů a patří mezi její diagnostická kritéria [82,83]. Nádor je obvykle velmi pomalu rostoucí a v některých případech dokonce nejsou známky progrese během několika let sledování nebo jsou dokumentovány i spontánní regrese [84,85]. Na zcela opačné straně spektra je 5 % pacientů s leptomeningeálními metastázami [86]. Právě pacienti s NF I mají dlouhodobě lepší prognózu s podílem progredujících nádorů mezi 15–75 % [87,88]. Může být postižena jakákoliv část nervu od optického traktu, chiazmatu, intrakraniální, intrakanalikulární až po intraorbitální část, která je postižena ve 25–50 % případů [89]. Existuje několik klasifikačních systémů. Nejstarší systém publikovaný Dodgem et al v roce 1958 rozděluje tumory na typ I postihující optický nerv, typ II postihující chiazma a typ III postihující tractus opticus nebo okolní struktury vč. hypothalamu [90]. Přepracovaná Dodgeova klasifikace byla publikována v roce 2008 a mimo precizní topické lokalizace zahrnuje i další faktory, jako je přítomnost NF I nebo leptomeningeální diseminace [91]. Poslední užívaný klasifikační systém je TxVx škála McCulloughova-Epsteinova popisující nejen lokalizaci, ale i funkční stav zraku (tab. 1) [92]. Pro indikaci léčby je v praxi kromě lokalizace tumoru nutné vždy přihlédnout k přítomnosti NF I, věku pacienta, symptomům a riziku obstrukce likvorových cest. U neurofibromatózy můžeme diagnostikovat i oboustranné postižení. Klinická prezentace závisí na lokalizaci tumoru. Při postižení hypothalamu je možná klinická manifestace endokrinopatie (pubertas praecox, porucha růstu při nedostatku růstového hormonu) nebo obstrukční hydrocefalus s nitrolební hypertenzí při obstrukci třetí komory. Typické příznaky pro lokalizaci v oblasti Dodge I a II jsou progredující porucha vizu, pomalu progredující diskrétní exoftalmus, nystagmus a strabizmus. Naopak poruchy okohybných nervů s diplopií, papiledém nebo atrofie optiku jsou příznaky méně časté. Ve skupině pacientů do dvou roků věku tvoří asi 20 % všech intrakraniálních tumorů, nebývá spojen s NF I, a má horší prognózu s obecně agresivnějším chováním, stejně jako u pacientů starších 10 let. Typickou klinickou prezentací v této skupině může být nystagmus, ztráta fixace při poruše vizu a spasmus nutans (triáda symptomů – oscilace očních bulbů, pokyvování hlavy a torticollis). Pozdější příznaky tvoří symptomy obstrukčního hydrocefalu a diencefalický Russelův syndrom (progredující hubnutí, zvracení, bez známek poruch výživy s normálním vzrůstem). V MR obraze je obvykle gliom izointenzní v T1 váženém obraze a hyperintenzní v T2 váženém obraze. Zvyšování signálu po podání gadoliniového kontrastu je variabilní, kdy můžeme pozorovat enhancující cystické komponenty i solidní neenhancující části. V MR obraze zvláště u NF I můžeme také pozorovt tortuozní nebo zalomený průběh zbytnělého optiku v orbitě (obr. 7). Zatímco při postižení optiku dominuje solidní rozšíření nervu bez cyst, posteriorně uložené tumory naopak cystickou komponentu obsahují často [93]. MR obraz často nekoreluje s deteriorací zraku, kdy u stacionárních lézí se zrak může zhoršovat a naopak u progredujících nemusí být ovlivněn. Známky zvyšujícího se nebo nového enhancementu jsou zpravidla spojeny s progresí a horší prognózou [94]. Vzhledem k dynamice a dlouhodobému sledování je vhodné využívat volumetrické hodnocení velikosti nádoru [95,96]. V CT obraze kromě rozšíření optického nervu můžeme pozorovat i dilataci optického kanálku. Z celé skupiny pacientů se odhaduje, že pouze 25–35 % pacientů vyžaduje vstupně terapii [97,98]. Cílem terapie je prevence deteriorace zraku a dlouhodobá kontrola vlastního nádoru. V případě asymptomatických pacientů nebo pacientů s minimálními příznaky není zřejmý benefit terapie oproti observaci [33,35,99]. Doporučený interval sledování je šest měsíců s event. prodloužením na 12 měsíců u stabilních pacientů. Terapie je indikována, kromě akutního řešení případného obstrukčního hydrocefalu, u pacientů s radiologickou progresí nebo deteriorací zraku. Chemoterapie je v současné době metoda první volby u většiny pacientů. Nejčastěji je používán protokol obsahující vinkristin a carboplatinu v první linii léčby a protokol s bevacizumabem a irinotecanem v případě recidivy [10,11,98,100]. Chirurgická terapie je indikována u pacientů s lokalizací v oblasti optického nervu s progresivním exoftalmem, nekorigovatelnou bolestí s úplnou ztrátou nebo těžkou deteriorací zraku. Kromě kombinace resekce s enukleací je možné provést i resekci šetřící vlastní oční bulbus. U pacientů s chiazmatickými nebo hypothalamickými tumory je indikována subtotální resekce v případě výrazného supraselárního šíření. Volba přístupu závisí na směru šíření a rozsahu nádoru (pterionální, interhemisferický, subfrontální, subtemporální přístup) [101]. V případě hydrocefalu bývá zaváděn ventrikuloperitoneální nebo ventrikuloatriální zkrat. Pro drénování cystické složky lze využít také Ommaya rezervoár. Biopsie bývá indikována vzácně při atypické klinické a radiologické manifestaci mimo obvyklé věkové rozmezí [36]. Radioterapie je indikována u starších pacientů bez NF I se selháním chemoterapie, v případě metastazování nebo u inoperabilních nádorů. U pacientů mladších tří let je kontraindikována pro vysoké riziko deteriorace kognitivních funkcí [44,45]. V souvislosti s radioterapií je také dokumentováno riziko rozvoje endokrinních dysfunkcí, rozvoje moya-moya syndromu a vznik sekundárních malignit, a to především ve skupině pacientů s NF I [102,103]. Slibnější stran nežádoucích vedlejších účinků se jeví stereotaktická radiochirurgie gama nožem nebo protonovým paprskem [51,104–106]. Zatímco na jedné straně moderní léčba dosahuje dlouhodobého 10letého přežití mezi 80 a 96 %, na straně druhé v případě snahy o dlouhodobé uchování zraku stále selhává [97,99,107]. Obecně horší prognózou mají posteriorně uložené a primárně symptomatické tumory, pacienti mladší jednoho roku a starší 10 let a pacienti s histologicky verifikovaným pilomyxoidním astrocytomem.


Meningeom pochvy zrakového nervu
Meningeomy pochvy zrakového nervu tvoří asi 2 % všech orbitálních tumorů a 1–2 % všech meningeomů [77,108–110]. Tyto nádory vyrůstající z obalu vlastního optického nervu je nutné odlišit od sekundárních meningeomů vrůstajících do orbity z okolí (např. sfenoorbitální meningeomy). Oddělit je nutno také zcela raritní skupinu tzv. ektopických volných meningeomů nesouvisející ani s kostěnou orbitou ani s vlastním obalem optiku [111]. Meningeom se může vyskytovat v celém průběhu optické pochvy od vlastního bulbu až po intrakraniální konec optického kanálu. Většina tumorů postihuje intraorbitální část a pouze menší část (8 %) tumorů má intrakanalikulární lokalizaci [109]. V některých případech může být infiltrace vlastního optiku i bulbu. Ke klasifikaci podle polohy a šíření nádorů lze využít navržený systém prezentovaný Schickovou (tab. 2, obr. 8.) [112]. Meningeom se pouze v 5 % vyskytuje bilaterálně a v tomto případě dominuje lokalizace intrakanalikulární [113,114]. Typické je jejich šíření cirkulárně kolem zrakového nervu. Vzhledem k tomuto intimnímu vztahu je ovlivněno vlastní piální cévní zásobení optiku a taktéž axonální transport s odpovídající klinickou prezentací [50,115–118]. Dřívější názor vycházející z peroperačních pozorování o možné primární neexistenci subarachnoidálních vrstvy v kanaliku-lárním úseku znemožňující resekci meningeomu byl vyvrácen elektron-mikroskopickými studiemi [119]. Histologicky se neliší od ostatních intrakraniálních meningeomů, nejčastěji jsou to přechodné (smíšené) nebo meningoteliomatozní typy [50]. Ženy jsou postiženy 3–5krát častěji než muži [109]. Průměrný věk je 40,8 roků (muži 36,1; ženy 42,5) [42,109]. Jako u ostatních meningeomů je v souvislosti s přítomností hormonálních receptorů popisována možná progrese v období těhotenství. V dětském věku bez dominance ženského pohlaví se vyskytuje 4–7 % nádorů. V této věkové skupině jsou tumory agresivnější, s vyšším rizikem intrakraniální a kontralaterální progrese, vyšším podílem recidiv a často jsou spojeny s neurofibromatózou typu II [9,109,115,120]. Klinicky se projevuje nádor nejdříve a nejčastěji pomalu progredující poruchou zrakové ostrosti a výpadky zrakového pole. Dalším příznakem zpravidla s delší latencí je pomalu progredující exoftalmus a porucha motility bulbu způsobená mechanicky nádorovou masou. Orbitální bolest bývá v různé míře až u poloviny pacientů [9,115,121,122]. Typická patognomická Walshova triáda s poruchou zraku, atrofií optiku a přítomností optociliární cévních zkratů způsobených chronickou kompresí centrální retinální žíly je přítomna pouze u menší části pacientů [9,115,121–123]. Dalšími příznaky může být papiledém, otok víček nebo chemóza spojivky. V radiologickém obraze na rozdíl od gliomů optiku není přítomna dilatace optického kanálu [111]. V CT obraze, kromě ztluštění optiku a homogenně se zvyšující denzity po podání kontrastní látky, můžeme pozorovat v 25–50 % případů kalcifikace [20,50,123]. Typický příznak tzv. tramvajových kolejí („tram track sign“) je způsoben dvěma hyperdenzními proužky nádoru oddělenými hypodenzním optickým nervem [109]. Základním vyšetřením jsou MR sekvence s potlačením tuku a podáním kontrastu, které mohou odhalit i drobné intrakanalikulární tumory a umožňují velmi dobře posoudit hranice nádoru [124]. Na T1 vážených sekvencích je nádor izointenzní nebo mírně hypointenzní v porovnání s mozkem a optickým nervem. Na T2 vážených sekvencích je typicky hyperintenzní, ale jsou popisovány i hypointezní varianty [50]. Opět je možné pozorovat na axiálních řezech „tram track sign“ a na koronárních řezech jeho obdobu tzv. „doughnut sign“ (příznak americké koblihy) s hypointenzním optikem obklopeným cirkulárním enhancujícím nádorem [20,42,123,125]. Klinický průběh je charakterizován pomalu během několika let progredující poruchou zraku s terminální slepotou postiženého oka event. obou očí při propagaci kontralaterálně a do oblasti chiazmatu [113,114]. Možné je identifikovat i ataky rychlého horšení zrakových funkcí. Na druhé straně existuje skupina pacientů s dlouhodobě stabilními zrakovými funkcemi během několikaleté observace a jsou dokumentovány i případy spontánního zlepšení [34]. Většina klinických sérií dokumentuje postupnou deterioraci zraku u 69–86 % pacientů [34,109,116,126].Saeed et al stanovili každoroční objemový přírůstek u nekalcifikovaných tumorů 23,5 mm3 a u kalcifikovaných 3,4 mm3 [127]. Ostatní studie prognostický faktor kalcifikací nepotvrdily. Konzervativní postup u stabilních pacientů musí kromě pravidelných MR kontrol obsahovat i ve stejném intervalu kontrolní oftalmologické vyšetření. Doporučovaný interval je pro první 2–3 roky šest měsíců, v případě dětí vzhledem k vyšší agresivitě nádoru ještě kratší [9,128]. Problémem progredující poruchy zraku je v čase se snižující šance na její zlepšení po terapii [9]. Výsledky chirurgické terapie jsou negativně ovlivněny nezbytnou deteriorací cévního zásobení optiku při vlastní resekci tumoru. Kompletní obalení optiku nádorem a šíření do anulus tendineus znemožňuje kompletní resekci a uchování zraku. Pouze vzácné případy se zlepšením vizu po chirurgické resekci jsou popsány v literatuře [112,116,127,129–131]. Nejen tyto subtotální a parciální resekce, ale i pouhé biopsie jsou doprovázeny vysokým rizikem komplikací se zhoršením zraku (30–80 %) nebo i jeho úplnou ztrátou při vysokém riziku další progrese rezidua (18–75 %) [9,109,110,112,115, 116,121,126,132]. Ani technika pouhé dekomprese optiku z podélné fenestrace postiženého obalu nevede ke zlepšení zraku, ale často opět k dalšímu zhoršení [9,50,115,117]. Chirurgická terapie je v současné době indikována u pacientů z těžkou poruchou anebo ztrátou zraku, při intrakraniálním šíření, při ohrožení chiazmatu a kontralaterálního optiku nebo při disfigurujícímu exoftalmu. V případě retrobulbárního postižení je prováděna resekce postiženého úseku nejčastěji z laterální orbitotomie a orbitopterionální kraniotomie. Kompletní revize optického kanálku až intrakraniálně s resekcí předního klinoideálního výběžku je součástí výkonu při intrakanalikulárním šíření. V případě vrůstání tumoru do očního bulbu je resekce rozšířena o enukleaci (obr. 4) [109,112,116]. Indikací k chirurgické akutní parciální resekci může být i vzácnější rychle progredující deteriorace zraku. Kombinace s pooperační radioterapií může dle několika kazuistik zrak zachránit [50,131]. Kombinovaná strategie s tzv. bezpečnou parciální resekcí a následnou radioterapií se svými komplikacemi neliší od skupiny čistě chirurgické [9,42,115,128,133]. Zatímco v případě chemoterapie nebyl prokázán benefit, radioterapie se stala vzhledem ke svým výsledkům metodou volby v terapii meningeomů pochvy optického nervu a nahradila dříve dominantní chirurgickou léčbu [8,9,12,47–50,121,134]. Terapie samostatnou frakcionovanou radioterapií nebo stereotaktickou radiochirurgií přináší nejlepší dlouhodobé výsledky stran zlepšení a dlouhodobého uchování zraku. Zlepšení je udáváno asi u 80 % pacientů, zatímco zhoršení jen asi u 10 % při dlouhodobé lokální kontrole kolem 95 % [9,12,42,46,48,49]. Dřívější vysoký výskyt komplikací až v 33 % v souvislosti s konvenčními radioterapeutickými technikami (poradiační retinopatie, okluze retinální arterie, perzistující iritida, atrofie temporálního laloku) [42,50] se podařilo moderními radioterapeutickými metodami snížit pod 10 % [9,50]. Většina z těchto komplikací je přechodných (cefalea, nauzea, fokální erytém a alopecie, otok), ale je třeba pacienta poučit i o možnosti vzniku vzácnějších závažnějších a pozdních komplikací (suché oko, retinopatie, optická neuropatie, hypofyzární dysfunkce, katarakta, poradiační onemocnění drobných cév mozku) [42,50]. V dnešní době se preferují moderní techniky, jako je stereotaktická frakcionovaná radiochirurgie, intenzitou modulovaná radioterapie, stereotaktická frakcionovaná radioterapie a 3D konformní radioterapie [9,12,48,49,109,115,121,126,128]. Z hlediska zraku lepší výsledky oproti jednorázovému radiochirurgickému ošetření gama nožem vykazují techniky frakcionované radiochirurgie s podobnými výsledky lokální kontroly nádoru [43,47]. Otázkou zůstává, kdy pacienta bez poruchy zraku nebo pouze s mírnou poruchou indikovat k terapii.


Nádory periferních nervů
Nádory periferních nervů tvoří 4–15 % všech orbitálních tumorů [18,135]. Do této skupiny zahrnujeme orbitální neurofibromy, schwannomy a maligní tumory z pochev periferních nervů (MPNST). Někteří autoři do této skupiny zařazují i vzácné amputační neuromy, které však nejsou problematikou onkologickou, ale jedná se o přirozenou posttraumatickou reakci přerušeného nervu u dříve prováděných neurektomií v orbitě, po enukleacích a vzácně i po operacích strabizmu [14,136].
Neurofibromy se vyskytují celkem ve třech variantách jako solitární, difuzní a plexiformní. Jedná se o benigní, pomalu rostoucí tumory složené z fibroblastů, Schwannových buněk a axonů [20]. Nejčastější orbitální forma je plexiformní neurofibrom, který je patognomický pro Recklinghausenovu neurofibromatózu NF I, a může se tudíž vyskytovat spolu s gliomem optiku. Typicky je popisován u dětí s predilekcí extrakonálně v oblasti horního laterálního kvadrantu a víčka. Patrná je deformace víčka a později šíření do okolních oblastí vč. temporální krajiny. Deformita je označována jako „bag of warms“ neboli pytel červů a typicky postupně esovitě zmenšuje oční štěrbinu a dislokuje bulbus inferiorně. Na MR je typicky hyperintenzní v T2 vážených sekvencích a heterogenní intenzity v T1 vážených obrazech s heterogenním zvyšováním intenzity po podání gadolinia [91]. Vzhledem k difuznímu šíření mezi tkáňovými vrstvami je chirurgická terapie paliativní s opakovanými resekcemi se snahou o zmenšení masy nádoru a zachování zrakových funkcí. V pozdějších fázích pak přichází v úvahu i exenterace. Vzácnější difuzní neurofibromy jsou méně často spojeny s NF I a difuzně extenzivně se šíří orbitou nejen tukem, ale i okohybnými svaly. Radikální resekce neohraničených tumorů je nemožná a opět zpravidla paliativní s časnými recidivami. Oba tyto typy neurofibromů jsou pokládány za prekurzor maligního tumoru z pochev periferních nervů s nejasným vlivem radioterapie, která bývá u terapie opakovaných recidiv využívána [137]. Solitární neurofibromy jsou v orbitě vzácné a vyskytují se ve vyšším věku (mezi 30. a 50. rokem) zpravidla opět v horním laterálním kvadrantu extrakonálně [77,138]. Ve 12 % jsou součástí NF I. Ačkoliv na rozdíl od schwannomů nemají pouzdro, jsou také velmi dobře ohraničeny a jakási pseudokapsula zjednodušuje jejich radikální resekci s velmi dobrou prognózou, bez nutnosti doplnění radioterapie. V klinickém i radiologickém obraze nelze solitární neurofibrom od schwannomu spolehlivě odlišit, kromě přítomnosti cyst, které jsou u neurofibromu vzácné [138].
Schwannomy jsou benigní pomalu progredující opouzdřené nádory podmíněné proliferací Schwannových buněk. Většinou jsou extrakonální z větví trigeminu, nejčastěji z frontální větve oftalmického nervu v horním kvadrantu, avšak mohou vyrůstat ze všech okohybných nervů, parasympatických a sympatických vláken, ale i ciliárního ganglia intrakonálně [77,135,139–143]. Histologicky se v rámci nádoru rozlišují alternující typy fascikulární Antoni A a retikulární typ Antoni B [139]. Vyskytují se v širokém věkovém rozmezí (20–70 let) a projevují se nejčastěji pomalu progredujícím exoftalmem a později i možnou poruchou vizu. Při postižení trigeminálních větví se může objevit hypestezie, dysestezie nebo bolest v inervované oblasti [141]. Při MR vyšetření se v T1 váženém obraze zobrazuje jako uniformně izointenzní velmi dobře ohraničený útvar s heterogenním zvýšením intenzity po podání kontrastní látky. V T2 váženém obraze je heterogenně hyperintenzní [91,139,140]. Při prorůstání horní orbitální štěrbinou může mít tvar přesýpacích hodin [139]. Chirurgická radikální resekce je u symptomatických schwannomů metoda volby s vynikající prognózou [140]. Stejně jako u ostatních schwannomů je stereotaktická radiochirurgie alternativou [51,144–147]. Vzácně může být součástí NF II a extrémně vzácná je i malignizace [148].
Maligní tumor z pochev periferních nervů (maligní schwannom, neurosarkom; MPNST) je vzácný agresivní nádor postihující hlavové nervy a orbitu. Bylo popsáno 35 případů postižení hlavových nervů, z toho kolem 15 případů v orbitě [149–157]. Radiologicky není odlišný od plexiformního nebo difuzního neurofibromu, avšak bývá přítomna destrukce skeletu orbity a měkkých tkání, propagace intrakraniálně a velmi rychlá progrese v porovnání s benigními tumory [91,137]. Nádor může vzniknout de novo (50 %) nebo upgradem neurofibromu a schwannomu. Asi 50 % nádorů se vyskytuje u NF I, s rizikem 2–29 % rozvoje během života [158,159]. V 39 % MPNST metastazuje (nejčastěji do plic) a 68 % pacientů na tumor zemře. Délka přežití je velmi variabilní mezi jedním měsícem a 23 lety při průměrném přežití dvou let 41 % a čtyř let 18 % [159–161]. Metoda volby je široká radikální resekce s následnou radioterapií [149,156,160,161]. Prognosticky negativní faktory jsou přítomnost NF I, velikost větší než 5 cm a inkompletní resekce [160]. V případech inkompletních resekcí, recidiv nebo generalizace se doplňuje chemoterapie v protokolu CYVADIC (doxorubicin, prokarbazin, cyklofosfamid, vinkristin) [161].
Angiogenní expanze
Problém většiny klasifikací orbitálních tumorů je mísení skutečných nádorů a nenádorových lézí, které by se měly zahrnovat do pseudotumorů. Právě v případě lézí cévního původu jsou do této skupiny zařazovány nesystematicky kavernózní hemangiomy, kapilární hemangiomy, lymfangiomy, hemangiopericytomy, stejně tak jako arteriovenózní malformace nebo karotido-kavernózní píštěle [18–20,74,77,162,163]. Cévní léze společně tvoří asi 12–15% orbitálních expanzí [19].
Kavernomy (kavernózní malformace, kavernózní hemangiomy), ačkoli nejde o skutečné nádory, ale benigní cévní velmi dobře ohraničenou nenádorovou nízkoprůtokovou malformaci s ektatickými, částečně trombozovanými a septovanými sinusoidálními venózními endotelem vystlanými kanály, jsou ve většině souborů uváděny jako nejhojnější benigní primární tumor orbity v dospělé populaci [18,20,77,164]. Nejčastěji se vyskytují od druhé do páté dekády věku a mohou mít sporadický i familiární výskyt [165,166]. Vyšší incidence u ženského pohlaví je dávána do spojitosti s expresí progesteronových receptorů u orbitálních kavernomů [167]. Kavernomy jsou lokalizovány obvykle intrakonálně, často v oblasti orbitálního apexu a většinou laterálně a projevují se velmi pomalu progredujícím exoftalmem (obr. 9) [168,169]. Pozdními příznaky jsou poruchy motility bulbu a až v pozdních fázích deteriorace zrakové ostrosti [165,170]. V MR obraze se zobrazují jako ostře ohraničené léze hypointenzní v T1 vážených sekvencích a hyperintenzní v T2 vážených sekvencích bez „flow void“ fenoménů [20,163,171]. Při trombóze může být signál v T1 sekvencích vyšší. Na CT můžeme identifikovat v některých případech kalcifikace a v pozdějších fázích remodelaci kostěné orbity [165,170,172,173]. Po podání kontrastní látky je typické pozdní zvyšování denzity odpovídající nízkému průtoku bez přímého arteriálního zásobení. Angiografický nález je typicky jako u intrakraniálních kavernomů negativní. V současné době díky dostupnosti radiologických vyšetření vzrůstá záchyt asymptomatických kavernomů [21,77]. Vzhledem ke klinickému průběhu s minimálním rizikem deteriorace zraku po spontánním drobném krvácení je primární volba terapie observace [171,174,175]. Chirurgická resekce je indikována až při rozvoji a progresi poruchy zraku, motility bulbu nebo progredujícím exoftalmu [18,163,174]. Po radikální resekci kavernomy nerecidivují, dokonce i parciální resekce symptomatického kavernomu může přinést vynikající dlouhodobý výsledek [168,170]. Alternativní terapie je jednorázová nebo frakcionovaná stereoradiochirurgie, kdy na rozdíl od intracerebrálních kavernomů je dokládána statisticky významná redukce objemu s regresí klinických příznaků podporující teorii o možné odlišnosti orbitálních a cerebrálních kavernomů [43,176,177].

Kapilární hemangiomy tvoří asi 3 % orbitálních expanzí a obvykle se prezentují u dětí již kolem šesti měsíců věku, jsou to benigní široce infiltrativní léze často postihující oční víčka s možnou kožní manifestací formou prokrveného „jahodového“ névu [19,77]. V dětské populaci se jedná o nejčastější angiogenní lézi v orbitě [74–76,178]. Častější jsou u chlapců 3 : 2 [74,178]. Mají po iniciální progresi a fázi stabilizace tendenci ke spontánní regresi během 3–5 let, ale mohou přetrvávat s významným kosmetickým deficitem. V případě nutnosti aktivní terapie se využívá pozitivní reakce na topické, intralezionální nebo celkové podávání kortikoidů a terapie laserem (argon, CO2) [179,180]. Pouze menší ohraničené ventrálně uložené kapilární hemangiomy je možné kompletně a bezpečně řešit excizí [19,77]. Pro refrakterní případy se používá interferon alfa [74]. Efekt mohou mít i betablokátory [181].
Lymfangiom je pomalu rostoucí benigní lokálně agresivní léze tvořená lymfatickými prostory vystlaným endotelem, která tvoří asi 1–5 % orbitálních expanzí [74,77,182]. Zajímavostí je, že sama orbita nemá lymfatickou drenáž. Vyskytuje se většinou v první dekádě věku a na rozdíl od kapilárního hemangiomu nemá tendenci ke spontánní regresi [183,184]. Může být postižena spojivka a projevuje se jako mnoholaločnatý namodralý útvar nejčastěji ve vnitřním horním kvadrantu [185]. Další formy jsou orbitální a generalizovaná kombinovaná s postižením často i kontralaterálním. Může být ve formě Kasabachova-Merrittova syndromu spojen s mozkovou arteriovenózní malformací a trombocytopenií [186]. Většinou kromě kosmetického deficitu je bez dalších symptomů. Typické jsou akutní spontánní hemoragie do lymfangiomu s tvorbou tzv. čokoládových cyst na základě následné kolikvace krve [187]. K akutní progresi dochází také při infektech. Vzhledem k infiltrativnímu růstu není možné kromě menších lokalizovaných lézí radikálně expanzi resekovat. Chirurgie se omezuje na vnitřní dekompresi, parciální resekci a aspiraci cyst u symptomatických lézí. U intrakonálních lézí lze využít aktinoterapii [182]. Další možností léčby je sklerotizace cyst různými látkami od fibrinových lepidel až po cytostatika nebo podávání kortikosteroidů [182,188].
Hemangiopericytomy se vyskytují v mladším a středním věku bez predilekce pohlaví a tvoří kolem 0,8–3 % orbitálních nádorů [18,189,190]. Jedná se o nízce maligní vaskularizované invazivní tumory s pomalým růstem a rizikem metastazování v 12–45 % [19,191]. Jejich chování bývá nepředvídatelné. Vyrůstají z populace nediferencovaných mezenchymálních pericytárních buněk. Typicky jsou uloženy extrakonálně. V T1 vážených sekvencích je izointenzní a v T2 vážených sekvencích mírně hyperintenzní. Po aplikaci kontrastní látky výrazně zvyšuje nádor svoji intenzitu [189]. Klinický projev je jako u jiných expanzí progredující exoftalmus a později diplopie. Radikální resekce je metoda volby. Ke snížení peroperačního krvácení se využívá předoperační endovaskulární embolizace [192,193]. Riziko lokální recidivy je i přesto vysoké (15–40 %) a může ho snížit pooperační radioterapie, která je indikována u recidiv [21,189,192–194].
Mezenchymální nádory
Rabdomyosarkom je nejčastější maligní primární tumor orbity v dětském věku. Vyskytuje se obvykle do 16 let věku s průměrným věkem pacientů sedm let [21,74–76,195]. Chlapci jsou postiženi častěji než dívky (5 : 3). Do 10 let se objevuje embryonální typ, zatímco u větších dětí a dospívajících převládá alveolární typ. Nevyrůstá ze svalů, ale z pluripotentních nediferencovaných mezenchymálních buněk v orbitě. Klinicky se projevuje rychle v řádu dní progredujícím exoftalmem s edémem a zarudnutím [163]. Nejčastěji je lokalizován v horním nazálním kvadrantu. V CT obraze může být patrná destrukce kostěné orbity a invaze do měkkých tkání. V T1 váženém obraze je izointenzní s jasným enhancementem po podání kontrastní látky a hyperintenzní v T2 váženém obraze [19]. Mohou být patrná ložiska krvácení [18,195]. Předpokládaná diagnóza musí být potvrzena jehlovou nebo lépe transorbitální biopsií s dostatečným ziskem materiálu. Terapie tohoto senzitivního nádoru musí začít co nejdříve vzhledem k prevenci ztráty zraku a zamezení další extraorbitální progrese. Dříve prováděná maximální resekce části tumoru event. i exenterace není již po bioptické verifikaci vyžadována [196]. Pacienti bez rezidua po provedené operaci jsou léčeni pouze chemoterapií, zatímco u pacientů s mikroskopickým nebo významným reziduem je terapie rozšířena o aktinoterapii [197]. Prognóza se velmi zlepšila s tříletým přežitím kolísajícím mezi 80–94 % u pacientů bez metastáz [74,198,199]. Negativními prognostickými faktory jsou věk do jednoho roku, alveolární typ rabdomyosarkomu, metastázy a propagace mimo orbitu nejčastěji do oblasti vedlejších dutin nosních a nazofaryngu [196,197]. Bez těchto faktorů převyšuje dnes pětileté přežití 90 %.
Fibrózní histiocytom patří k častějším orbitálním tumorům v dospělé populaci s maximem výskytu v páté a šesté dekádě věku [18]. Nejčastější forma je benigní (65 %), která je dobře ohraničená. Lokálně agresivní forma (25 %) a forma maligní (10 %) jsou infiltrativní [200]. Maligní formy jsou popisovány v souvislosti s anamnézou aktinoterapie retinoblastomu, ale také po parciálních resekcích vlastního histiocytomu. Klinicky benigní forma se projevuje pomalu několik let progredujícím exoftalmem. Radiologicky se jeví jako ohraničená nebo infiltrativní léze s částečným enhancementem po podání kontrastní látky. Metoda volby je chirurgická radikální excize zabraňující riziku recidivy. Desetileté přežití je u benigní formy 100 %, u maligní i přes adjuvantní onkologickou terapii kolem 25 % [201].
V orbitě je možné vzácně zachytit také celou řadu dalších mezenchymálních nádorů, jako jsou fibrom, fribrosarkom, myofibrom, angiofibrom, solitarní fibrózní tumor, leiomyom, leiomyosarkom nebo lipomy a liposarkomy [19,77,162]. Přesné zařazení mezenchymálního tumoru lze provést až po pečlivé histologické analýze.
Lymfomy
Lymfoproliferativní nádory jsou nejčastější primární orbtitální tumory ve věkové skupině nad 60 let a nejčastější maligními tumory očnice v dospělém věku vůbec [21,202,203]. Lymfomy tvoří asi 90 % těchto expanzí a celkem 24 % orbitálních expanzí nad 60 let věku [202]. Lymfomy mohou být jak primární nejčastěji z buněk marginální zóny (non-hodgkinské MALT lymfomy), tak v případě orbity extranodální z populací lymfocytů v substantia propia spojivky nebo v intersticiální tkáni slzné žlázy [204]. Mezi ostatní lymfomy v orbitě patří další non-hodgkinské typy (difuzní velkobuněčný B lymfom, B lymfom z buněk pláště, folikulární B lymfom). Kromě primárních lymfomů se může jednat i o projev systémového onemocnění. Až v 30 % je popisován rozvoj systémového onemocnění po diagnóze primárního orbitálního lymfomu [202]. Většina lymfomů je unilaterálních (76 %) a uložených extrakonálně s postižením slzné žlázy asi v 40 % [20,202]. V MR obraze se ukazují jako difuzní neohraničené léze a v druhé polovině jako ohraničené expanze (obr. 10). Charakteristický je obraz „oblévání“ orbitálních struktur (bulbus, optický nerv, svaly). V T1 sekvencích je izointenzní a T2 sekvencích hyperintenzní s uniformním zvyšováním intenzity po podání kontrastní látky [205,206]. Typický je obraz restrikce difuze v difuzně vážených obrazech a ADC mapách MR obdobně jako u intrakraniálních lymfomů. Díky těmto sekvencím může být lymfom bezpečně odlišen od zánětlivého pseudotumoru [207]. Klinicky se projevují exoftalmem, patrnou vyklenující se masou tumoru, viditelnou subkonjunktivální komponentou a poruchou motility bulbu. Důležité je odlišení od zánětlivého pseudotumoru, který dříve byl označován jako benigní lymfom [208]. Nástup příznaků bývá u pseudotumoru akutní, je provázen bolestí a v MR obraze, kromě odlišnosti v difuzně vážených sekvencích, bývá infiltrace okohybných svalů [20]. V případě podezření na lymfom by mělo být u pacienta nejprve vyloučeno systémové postižení (punkce kostní dřeně, CT hrudníku a břicha) a teprve následně by se mělo přistoupit k biopsii orbity. Léčba je vedena hematoonkologem dle histologické typizace a stagingu kombinací chemoterapie (protokol CHOP) a aktinoterapie. Lokální kontrola po pěti letech je udávána s ohledem na použitou dávku při radioterapii na 80–100 % [209].

Nádory slzné žlázy
Expanze slzné žlázy tvoří asi 5–14 % primárních expanzí orbity [77,210]. Epiteliální nádorové procesy tvoří asi 40–50 %. Nonepiteliální procesy zahrnují zánětlivé postižení (dakryoadenitida) a v 50 % lymfoproliferativní nádory [211]. Naprostá většina těchto lymfoproliferativních nádorů jsou non-hodgkinské B lymfomy. V některých sestavách dokonce tvoří nejčastější nádorové léze slzné žlázy [77]. Typický je pro ně vyšší věk, nejčastěji sedmé decennium, a nebolestivá masa s progredujícím exoftalmem [20]. Terapie a radiologické charakteristiky se shodují s ostatními lymfomy.
Pleomorfní adenom (benigní smíšený nádor) je nejčastější benigní nádor slzné žlázy tvořící až 57 % epiteliálních nádorů [211]. Nádor je složen z mezenchymálních i epiteliálních elementů s tvorbou ohraničující pseudokapsuly. Nádor postihuje nejčastěji pacienty ve čtvrté a páté dekádě věku [212]. Klinicky se projevuje pomalu rostoucí nebolestivou masou v horním laterálním kvadrantu dislokující bulbus dolů a mediálně s „S“ tvarem horního víčka [21,213]. V CT obraze se jedná o dobře ohraničenou, homogenně enhancující lézi po podání kontrastní látky. Větší nádory mohou být heterogenní s cystickými degeneracemi, hemoragiemi, nekrózami a mukoidními kolekcemi [212]. Nejsou známky eroze kosti, ale hladká remodelace v místě fossa lacrimalis [20]. V MR obraze je nespecificky hypointenzní v T1 vážených sekvencích a hyperintenzní v T2 vážených sekvencích. Metodou volby v terapii je en bloc chirurgická resekce nejčastěji z laterální orbitotomie. Parciální resekce vedou k recidivám a možné malignizaci nádoru. Vyhýbáme se také biopsiím pro vysoké riziko implantačních metastáz [214]. V případě radikální resekce je prognóza excelentní [215].
Adenoidně cystický karcinom je nejčastější maligní epiteliální tumor slzné žlázy a tvoří asi 5 % všech primárních orbitálních tumorů [215]. Vrchol výskytu je ve čtvrté dekádě věku bez predilekce pohlaví [209]. Histologicky je popisováno několik subtypů s prognosticky nejhorším bazaloidním karcinomem, kdy pětileté přežití je pouze kolem 21 %, na rozdíl od ostatních subtypů, kde přesahuje 70 % [216]. V poměrně rychle se rozvíjejícím klinickém obraze je charakteristická, kromě masy tumoru v superolaterálním kvadrantu, přítomnost postupně se rozvíjející neuropatické bolesti z perineurální invaze nejčastěji podél větví oftalmického nervu. Na rozdíl od zánětlivých lézí však oko bývá klidné a bolest není zhoršována pohybem bulbu [21]. V CT obraze je možno identifikovat eroze kosti a častější jsou také kalcifikace. MR potvrdí infiltrativní růst, tvorbu nodulů mimo slznou žlázu a rovněž perineurální šíření [20]. Základem terapie je chirurgická resekce tumoru s event. exenterací očnice. Radikální resekce je limitována především propagací nádoru perineurálně a intrakraniálně. Limitující mohou být i vzdálené metastázy v plicích. Chirurgická léčba je doplňována radioterapií s iradiací kavernózního splavu, horní orbitální fissury a orbitálního apexu [209]. Celková prognóza není dobrá s 10letým přežitím pouze 20 % pacientů [211].
Sekundární orbitální nádory
Sekundární orbitální tumory primárně vznikají mimo orbitální prostor a teprve sekundárně se šíří do orbity z okolí. Přirozenou hranicí a také blokádou šíření nádorového procesu je periorbita. Na hranici mezi primárními a sekundárními nádory jsou primární tumory postihující kostěnou očnici.
Primární kostěné tumory jsou velmi heterogenní skupina a tvoří celkem 0,6–2 % všech orbitálních nádorů [184,217]. Nejčastějšími nádory tvořícími 35 % této skupiny jsou osteomy a fibrózní dysplazie [217]. Osteomy se typicky vyskytují v oblasti etmoidální, frontoetmoidální a frontální nejčastěji ve čtvrté a páté dekádě věku bez predilekce pohlaví. Většina osteomů je asymptomatická, při klinické manifestaci se projevuje velmi pomalu progredujícím exoftalmem a dislokací bulbu. Kromě přímé propagace osteomu do orbity se může projevit i sekundárně prostřednictvím mukokély při okluzi ostia frontálního sinu [217]. Terapie symptomatických osteomů je chirurgická. Fibrózní dysplazie se vyskytuje ve formě monoostotické, polyostotické či jako součást McCuneova-Albrightova syndromu a je pro ni charakteristická náhrada fyziologické kosti fibrózně-kostěnou tkání [217]. Nejčastěji v rámci monoostotické formy je postižena frontální, etmoidální nebo sfenoidální kost. Projevuje se pomalu progredující deformitou obličeje, exoftalmem, deviací bulbu a bolestí orbity nebo hlavy. Maligní transformace projevující se rychlou progresí příznaků je vzácná do 0,5 % případů a riziko se zvyšuje po proběhlé radioterapii až na 15 % [218]. Propagace do optického kanálku může způsobit poruchu zraku. V CT obraze je popisován tzv. pagetoidní vzhled se setřelou kresbou kosti a obrazem „mléčného skla“ (obr. 11). MR vyloučí i nevýznamnou měkkotkáňovou komponentu typickou pro sfenoorbitální meningeomy. Chirurgická terapie je rezervována pro pacienty s funkčním deficitem, nekorigovatelnou bolestí, významnou deformitou nebo podezřením na malignizaci. Preventivní dekomprese optického kanálku není doporučována vzhledem k tomu, že u většiny pacientů i s postižením optického kanálku se nerozvine porucha vizu [219,220]. Další možností terapie je nasazení bisfosfonátů, které na základě ovlivnění maturace a migrace osteoklastů pozitivně ovlivňují bolest a velikost ložiska dysplazie [221,222]. V oblasti kostěné orbity se může vzácně vyskytnout také osteoblastom, chondrom, aneuryzmatická kostní cysta, obrovskobuněčný kostní nádor, osteosarkom, chondrosarkom, Ewingův sarkom nebo intraoseální hemangiom [19,217]. Vzácný je také solitární plazmocytom orbity. Většinou je postižení diagnostikováno v rámci systémového mnohočetného myelomu [223].

Sfenoorbitální meningeomy jsou charakterizovány dominantní intraoseálním postižením křídla kosti klínové s nedominantní intrakraniální propagací formou ploché „en plaque“ měkkotkáňové složky [18,224]. Právě přítomnost měkkotkáňové složky odlišuje v MR obraze tyto meningeomy od fibrózní dysplazie. Rozsah postižení skeletu je nutné hodnotit z CT vyšetření v kostním okně. Obdobně jako u ostatních meningeomů je predilekce u žen a vrchol výskytu ve čtvrté a páté dekádě věku. V klinickém obraze dominuje pomalu progredující exoftalmus, později porucha zraku a diplopie. Porucha vizu je dána kompresí optiku, která může být nejen typicky v oblasti optického kanálku a apexu orbitu, ale i v oblasti intrakraniální [21]. Vlastní pochva optického nervu na rozdíl od meningeomu pochvy optického nervu není nádorem postižena. Léčba je indikována u symptomatických nebo progredujících meningeomů. Zatímco u laterálních variant je možno provézt resekci radikální, v případě mediálních variant je chirurgická terapie limitována postižením kavernózního splavu, horní orbitální fissury a oblasti apexu orbity. Resekce je prováděna některou z variant pterionálního přístupu s dekompresí optického kanálku, intradurální a intraorbitální revizí (obr. 12) [225,226]. Reziduální nádor lze ošetřit stereotaktickou radiochirurgií následovně po chirurgickém výkonu nebo vyčkat radiologické progrese [226]. Recidivy a progrese reziduí jsou relativně časté (27–50 %) [225]. Kromě sfenoorbitálních meningeomů se mohou propagovat do očnice i ostatní intrakraniální meningeomy v této oblasti.

Z dalších intrakraniálních tumorů byla popsána propagace do orbity u adenomů hypofýzy, kraniofaryngeomů nebo chordomů klivu [21]. Variantou propagace intrakraniálních nádorů do očnice je i šíření cestou subarachnoideálních prostorů kolem optického nervu např. u karcinomatózy mening a u lymfomů [21].
Dermoidy a epidermoidy, jež tvoří asi 2 % operovaných orbitálních tumorů, jsou benigní cystické choristomy vznikající z embryonálně dislokované ektopické ektodermální tkáně [76,77,184]. Dermoidy na rozdíl od epidermoidů obsahují i kožní adnexa vč. vlasů. U choristomů není predilekce pohlaví a častěji se vyskytuji v dětském věku, ale můžeme je diagnostikovat i v pozdějším věku. V dětském věku tvoří s 30–46 % nejčastěji operovanou orbitální expanzi [75,76]. V oblasti orbity je typická lokalizace frontozygomatická sutura, méně často je nalézáme v horním mediálním kvadrantu [227]. Růst je pomalý a je dán akumulací dermálních produktů. V případě lokalizace před orbitálním septem může být ložisko dobře hmatné, zatímco u posteriorně ložených se projeví až expanzivním chováním. Vzácněji může dojít úrazem nebo spontánně k ruptuře s lokální zánětlivou reakcí [74]. V radiologickém zobrazení odpovídá obsah obrazu tukové tkáně. V CT obraze jsou typické ostré hladké sklerotické hranice kostního defektu. V MR obraze je typická hyperintenzita v T1 vážených sekvencích a také vysoký signál v difuzně vážených sekvencích (obr. 13) [228]. Enhancement je limitován jen na pouzdro cysty. Metoda volby v terapii je u symptomatických lézí radikální chirurgická resekce s výbornou dlouhodobou prognózou. Parciální resekce nebo dokonce aspirace vedou k recidivám [229]. V případě lokální zánětlivé reakce podáváme kortikoidy.

Typická skupina sekundárních orbitálních nádorů jsou nádory přímo se šířící z vedlejších dutin nosních, obvykle prostřednictvím lokální eroze kosti, ale i preformovanými fyziologickými kanály [230,231]. Vedle benigních tumorů (invertovaný papilom, juvenilní angiofibrom) dominují malignity (adenokarcinom, adenoidně cystický a mukoepidermoidní karcinom, skvamózní karcinom, dediferencovaný karcinom, dlaždicový karcinom) (obr. 14) [230,231]. Vzácnější je maligní olfaktorický neuroblastom (estezioneuroblastom) vycházející z neuroektodermových olfaktoriálních buněk nosní sliznice. Vyskytuje se hlavně u dětí a mladistvých s postižením orbity až u 2/ 3 pacientů [21]. Celkem v oblasti vedlejších dutin nosních můžeme diferencovat kolem 70 histologických subtypů nádorů [13]. U pacientů se před příznaky orbitálními setkáváme s anamnézou chronické sinusitidy, nazální obstrukce a epistaxe. K dalším příznakům patří bolest a rinorea. Současně při intrakraniální propagaci se objevují neurologické příznaky z oblasti frontálních laloků [232]. Metastatické postižení krčních lymfatických uzlin se pohybuje v době diagnózy mezi 5 a 20 %, vzdálené hematogenní metastázy jsou vzácnější [232]. Metastázy, stejně jako postižení očnice, pterygomaxillární fossy, kavernózního splavu a intrakraniální propagace, jsou negativní prognostické faktory. Očnice je postižena téměř u čtvrtiny pacientů [232]. Metoda volby je kromě paliativní léčby v pokročilých stadiích chirurgická resekce s následnou radioterapií vč. ozáření spádových krčních lymfatických uzlin [232]. V poslední době se postupně upouští od mutilujících radikálních exenterací orbity [233] se snahou ji uchovat [234,235]. Poslední publikovaná metaanalýza tento trend podporuje bez průkazu benefitu exenterace na celkovou prognózu [13]. Všeobecně přijímané kritérium k indikaci exenterace je grade III dle Iannettiho s postižením svalů, optiku, bulbu nebo kůže (tab. 3) [236]. Celkové pětileté přežití se pohybuje mezi 20 a 90 % [13,232,233,236]. Orbitální extenze tumoru ve skupině estezioneuroblastomů neovliňuje celkové pětileté přežití (78 %), ale snižuje časový interval recidivy nádoru [237].


Sekundárně očnici postihují také nádory primárně lokalizované v bulbu (melanom, retinoblastom), spojivce (melanom) nebo v oblasti očních víček (bazaliom, spinocelulární karcinom) [21].
Metastázy
Kolem 1–13 % nádorů orbity tvoří metastázy, které mohou postihovat jak intraorbitální struktury, tak kostěnou očnici [20,77,162,238]. V dětském věku je nejčastější zdroj metastáz neuroblastom (až v 40 %) a Ewingův sarkom [19]. Typicky se objevuje rychle nastupující exoftalmus a doprovázející periokulární ekchymózy [74]. V případě neuroblastomu je bilaterální postižení až v 50 % [18]. Další zdroj metastáz u dětí a mladistvých jsou nádory varlat, histyocytóza a leukemie. V dospělé populaci jsou zdrojem metastáz typicky karcinomy prsu a plic [77]. Častěji než do orbity metastazují nádory v dospělosti intraokulárně do uvey (8 : 1). V orbitě tvoří až 50 % metastáz metastázy karcinomu prsu a z toho plyne predilekce ženského pohlaví [20]. Metastázy mohou být první příznak primárního onemocnění až v 25 % [19]. Mezi další maligní nádory metastazující do očnice patří nádory prostaty, gastrointestinální, štítnice, ledviny nebo melanom. Klinicky se projevují bolestivým rychle progredujícím exoftalmem, poruchou motility bulbu a destrukcí kostěné očnice. Vzácněji asi v 10 % se může v případě skirhotického karcinomu prsu projevit paradoxním enoftalmem, na podkladě fibrotického jizvení retrobulbárního tuku [239]. V MR obraze není nález specifický s hypointezitou v T1 váženém a hyperintezitou v T2 váženém obraze a enhancementem kontrastní látky. Zpravidla mají difuzní a infiltrativní charakter [20,163]. Metastázy jsou uloženy v 60 % extrakonálně, v 20 % intrakonálně a ve zbylých 20 % jsou difuzní [21]. Až v 30 % lze při MR odhalit současné metastatické postižení mozku. Terapie se odvíjí od primárního onemocnění, chemo - a radiosenzitivity primárního nádoru a především celkové prognózy. Aktivní přístup je volen u solitárních metastáz s celkovou dobrou prognózou pacienta. Léčba spočívá v kombinaci radikální nebo subtotální resekce a radioterapie event. chemoterapie. Medián přežití se pohybuje kolem 12 měsíců s dvouletým přežitím méně než v 30 % [238].
Pseudotumory
Do skupiny pseudotumorů zahrnujeme nesourodou skupinu expanzivních, nádorům podobných lézí, které však nemají původ nádorový a je třeba je diferenciálně diagnosticky odlišit od skutečných nádorů.
Mukokély jsou cystické, pomalu expanzivní léze vznikající ve vedlejších dutinách nosních při obstrukci přirozeného ústí dutin na základě stagnace mucinózního sekretu a chronického tlaku na okolní kostěné hranice. Nejčastěji se objevují ve frontálním a etmoidálních sinech. Projevují se pomalu progredující dislokací bulbu a defigurací orbitální hrany [18]. V CT obraze jsou homogenně izodenzní s jasně definovanými hranicemi osteolýzy s možnou sklerotizací okrajů [21]. MR obraz se mění v závislosti na stáří a obsahu mukokély [18]. Léčba je chirurgická s obnovením fyziologické drenáže dutiny (obr. 15). Dříve používané zevní přístupy s resekcí stěny cysty byly nahrazeny endoskopickými fenestracemi s uchováním přirozené výstelky [240–242].

Orbitální encefalokély jsou způsobeny prolapsem intrakraniálního obsahu kongenitálním nebo postraumatickým defektem ve stropu očnice [243]. Vzniklý exoftalmus mívá pulzatilní charakter podmíněný přenosem intrakraniální pulzace likvoru. V případě kongenitální encefalokély chybí anamnéza traumatu, její lokalizace je vázána ve střední čáru a bývá patrný hypertelorizmus. Kongenitální orbitální encefalokély jsou velmi vzácné a patří do skupiny bazálních a nazoorbitálních encefalokél [244]. Posttraumatické encefalokély se mohou manifestovat i mnoho let po iniciálním úrazu a jsou častější u dětí [245]. Terapie je chirurgická s resekcí dysplastické a gliotické mozkové tkáně herniované do očnice, plastikou tvrdé pleny a rekonstrukcí stropu očnice [243–245].
Gravesova endokrinní oftalmopatie je nejčastější příčina bilaterálního nebo vzácně unilaterálního (10–15 %) exoftalmu u dospělých, především žen středního věku [246]. Onemocnění je autoimunitního charakteru s vazbou protilátek na buňky štítné žlázy, okohybných svalů a pojivové tkáně orbitálního tuku. Kromě exoftalmu je typická retrakce horního víčka a porucha motility bulbu. Na rozdíl od zánětlivého pseudotumoru nebývá přítomna bolest. V endokrinologickém obraze je v 75 % patrná hypertyreóza se zvýšením T3 a T4 se snížením TSH. V MR obraze je typické vřetenovité ztluštění okohybných svalů typicky inferiorního a mediálního, bez postižení jejich úponů a s absencí orbitální masy [206]. Metoda volby je podávání vysokých dávek kortikoidů a event. jiných imunosupresiv [246]. U akutní deteriorace zraku bez odpovědi na kortikoterapii je indikována radioterapie a event. dekompresní operace orbity [18,209]. K chirurgické dekompresi bývá indikováno jen asi 5–6 % pacientů nereagujících na konzervativní terapii a vzácněji pacienti z kosmetické indikace. Operace lze rozdělit na výkony vedoucí k dekompresi kostěného skeletu očnice s otevřením periorbity, zákroky odstraňující část orbitálního tuku a též jejich vzájemné kombinace. Byla popsána celá řada operačních dekompresních technik, mezi které patří: laterální dekomprese (Dollinger 1911), transfrontální dekomprese stropu (Naffziger 1931), mediální dekomprese (Sewall 1936), dekomprese spodiny (Hirsch 1950), transantrální dekomprese spodiny a mediální stěny (Walsh, Ogura 1957) a endonazální apikální dekomprese (Kennedy 1990, Michel 2001) [247]. V současné době nejsou k dispozici dostatečná data k preferování některé z dekompresních technik [248,249]. Takzvaná balancovaná současná dekomprese laterální a mediální stěny očnice eliminující jednostrannou deviaci polohy očního bulbu patří k nejlépe hodnoceným z hlediska efektu a výskytu komplikací (diplopie, porucha motility bulbu) [250].
Zánětlivý pseudotumor (nespecifická myositis, idiopatický zánět očnice) je imunitně podmíněný neinfekční zánět nejasného původu postihující kteroukoliv měkkou tkáň v očnici, jenž patří mezi poměrně časté příčiny exoftalmu [204,251]. Afekce bývá častěji jednostranná s výraznou bolestí, exoftalmem, chemózou spojivky, poruchou hybnosti bulbu, edémem a zčervenání víček, ale bez celkových příznaků a febrilií [18]. U dospělých se může vyskytnout také forma lokalizovaná, nebolestivá na celkově klidném oku. V případě lokalizace v hrotu očnice, horní orbitální štěrbině nebo kavernózním splavu se může projevit jako Tolosův-Huntův syndrom bolestivé oftalmoplegie [18,252]. V histologii, která je v případě dostupnosti procesu odebrána biopsií, je nespecifická směs akutního a chronického zánětu s eozinofily, plazmocyty a lymfocyty, avšak bez infekčního agens. V MR obraze na rozdíl od endokrinní orbitopatie proces infiltruje sval vč. šlachy s typickým postižením mediálního a laterálního přímého svalu [18,206]. V diferenciální diagnostice je třeba myslet na metastázy, lymfom, rabdomyosarkom, infekční záněty a systémová onemocnění (sarkoidóza) [252]. Terapie volby je pulzní aplikace velkých dávek kortikoidů, která se využívá také jako diagnostický test. Příznaky regredují rychle v řádech dnů. V případech s nedostatečnou odezvou na kortikoterapii rozšiřujeme léčbu o cytostatika, radioterapii, event. u chirurgicky přístupných lézí (nodulární dakryoadenitida) o excizi [18,204, 209,252]. Udržovací léčba perorálnímí kortikokoidy trvá několik měsíců a její předčasné ukončení zvyšuje riziko recidivy [252].
Závěr
Nádory očnice tvoří relativně vzácnou a velmi heterogenní skupinu patologií se stejně heterogenními terapeutickými postupy. Optimální je léčba vedená prostřednictvím multidisciplinárních týmů ve specializovaných centrech zabývajících se touto problematikou.
Poděkování patří Martině Vlčkové za rešeršní a meziknihovní služby.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 1. 9. 2015
Přijato do tisku: 16. 11. 2015
MUDr. Petr Vachata, Ph.D.
Neurochirurgická klinika
UJEP a Krajská zdravotní a.s.
Masarykova nemocnice v Ústí nad Labem, o.z.
Sociální péče 12A
401 13 Ústí nad Labem
e-mail: vachata@gmail.com
MUDr. Petr Vachata, Ph.D.

Promoval na lékařské fakultě v Hradci Králové v roce 1998 s vyznamenáním a cenou děkana lékařské fakulty pro nejlepší absolventy. Od promoce pracuje na Neurochirurgické klinice v Ústí nad Labem pod vedením prof. Sameše, s nímž založil první anatomickou disekční laboratoř v České republice. Práce této laboratoře a pravidelné pořádání mezinárodních disekčních kurzů mimo jiné vedlo Světovou neurochirurgickou společnost k zařazení pracoviště jako jediného ve střední Evropě mezi centra chirurgie lební baze kategorie A. V roce 2010 obhájil dizertační práci v oboru neurověd pod vedením školitele prof. Petrovického na téma „Mikrochirurgická anatomie transtemporálních přístupů k lební bazi“. Je autorem a spoluautorem 54 odborných článků v domácích i zahraničních časopisech a monografiích. Podílel se na učebnici neurochirurgie, která byla oceněna Českou neurochirurgickou společností. Pravidelně přednáší doma i v zahraničí. Je spoluřešitelem čtyř grantových projektů. V roce 2001 získal fellowship Evropské pediatrické neurochirurgické společnosti a opakovaně v letech 2005 a 2007 fellowship Evropské společnosti chirurgie lební baze. Stážoval na pracovištích prof. Sena (New York) a prof. Bertallanfyho (Marburg). Věnuje se celému rozsahu neurochirurgie s klinickým zaměřením na chirurgii lební baze, cévní neurochirurgii, spondylochirurgii a dětskou neurochirurgii. Pravidelně se účastní jako školitel mezinárodních disekčních kurzů.
Sources
1. Rubin PA, Remulla HD. Surgical methods and approaches in the treatment of orbital disease. Neuroimaging Clin N Am 1996; 6(1): 239–255.
2. Knapp H. A case of carcinoma of the outer sheath of the optic nerve, removed with preservation of the eyeball. Arch Ophthalmol Otol 1874; 106 : 323–354.
3. Kronlein RU. Zur pathologie und operativen behandlung der dermoidcysten der orbita. Beitr Klin Chir 1889; 4 : 149–163.
4. Dandy WE. Results following the transcranial operative attack on orbital tumors. Arch Ophthalmol 1941; 25 : 191–216.
5. Frazier CH. An approach to the hypophysis through the anterior cranial fossa. Ann Surg 1913; 57(2): 145–150.
6. Norris JL, Cleasby GW. Endoscopic orbital surgery. Am J Ophthalmol 1981; 91(2): 249–252.
7. Taveras JL, Haik BG. Radiography of the eye and orbit: a historical overview. Surv Ophthalmol 1988; 32(5): 361–368.
8. Becker G, Jeremic B, Pitz S, Buchgeister M, Wilhelm H, Schiefer U et al. Stereotactic fractionated radiotherapy in patients with optic nerve sheath meningioma. Int J Radiat Oncol Biol Phys 2002; 54(5): 1422–1429.
9. Berman D, Miller NR. New concepts in the management of optic nerve sheath meningiomas. Ann Acad Med Singapore 2006; 35(3): 168–174.
10. Packer RJ, Ater J, Allen J, Phillips P, Geyer R, Nicholson HS et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg 1997; 86(5): 747–754.
11. Packer RJ, Jakacki R, Horn M, Rood B, Vezina G, MacDonald T et al. Objective response of multiply recurrent low-grade gliomas to bevacizumab and irinotecan. Pediatr Blood Cancer 2009; 52(7): 791–795. doi: 10.1002/ pbc.21935.
12. Pitz S, Becker G, Schiefer U, Wilhelm H, Jeremic B, Bamberg M et al. Stereotactic fractionated irradiation of optic nerve sheath meningioma: a new treatment alternative. Br J Ophthalmol 2002; 86(11): 1265–1268.
13. Reyes C, Mason E, Solares CA, Bush C, Carrau R.To preserve or not to preserve the orbit in paranasal sinus neoplasms: a meta-analysis. J Neurol Surg BSkull Base 2015; 76(2): 122–128. doi: 10.1055/ s-0034-1390403.
14. Otradovec J. Choroby očnice. Praha: Avicenum 1986.
15. Zide BM, Jelks GW. Surgical anatomy of the orbit. 1st ed. New York: Raven Press 1985.
16. Rhoton AL jr, Natori Y. The orbit and sellar region: microsurgical anatomy and operative approaches. 1st ed. New York: Thieme Medical 1996.
17. Dutton JJ. Atlas of clinical and surgical orbital anatomy. 2nd ed. London: Elsevier Saunders 2011.
18. Bejjani G, Maroon JC, Kennerdell JS. Orbital tumors. Philadelphia: Saunders 2001.
19. Darsaut TE, Lanzino G, Lopes MB, Newman S. An introductory overview of orbital tumors. Neurosurg Focus 2001; 10(5): 1–9.
20. Tailor TD, Gupta D, Dalley RW, Keene CD, Anzai Y. Orbital neoplasms in adults: clinical, radiologic, and pathologic review. Radiographics 2013; 33(6): 1739–1758. doi: 10.1148/ rg.336135502.
21. Volpe NJ, Gausas RE. Optic nerve and orbital tumors. Neurosurg Clin N Am 1999; 10(4): 699–715.
22. Howard GM. The orbit. Arch Ophthalmol 1968; 80(5): 667–680.
23. Khan SN, Sepahdari AR. Orbital masses: CT and MRI of common vascular lesions, benign tumors, and malignancies. Saudi J Ophthalmol 2012; 26(4): 373–383. doi: 10.1016/ j.sjopt.2012.08.001.
24. Peeters F, Kröger R, Verbeeten B jr, Verteege C. The value of computer tomography, orbital venography and carotid angiography in the diagnosis of exophthalmos. Radiol Clin (Basel) 1977; 46(6): 430–438.
25. Weisman RA, Kikkawa D, Moe KS, Osquthorpe JD. Orbital tumors. Otolaryngol Clin North Am 2001; 34(6): 1157–1174.
26. Sirin S, Schlamann M, Metz KA, Bornfeld N, Schweiger B, Holdt M et al. High-resolution MRI using orbit surface coils for the evaluation of metastatic risk factors in 143 children with retinoblastoma: Part 1: MRI vs. histopathology. Neuroradiology 2015; 57(8): 805–814. doi: 10.1007/ s00234-015-1544-2.
27. Sirin S, Schlamann M, Metz KA, Bornfeld N, Schweiger B, Holdt M et al. High-resolution MRI using orbit surface coils for the evaluation of metastatic risk factors in 143 children with retinoblastoma: Part 2: new vs. old imaging concept. Neuroradiology 2015; 57(8): 815–824. doi: 10.1007/ s00234-015-1538-0.
28. Bangiyev L, Raz E, Block TK, Hagiwara M, Wu X, Yu E et al. Evaluation of the orbit using contrast-enhanced radial 3D fat-suppressed T weighted gradient echo (Radial-VIBE) sequence. Br J Radiol 2015; 88(1054): 20140863. doi: 10.1259/ bjr.20140863.
29. Tsiapa I, Tsilimbaris MK, Papadaki E, Bouziotis P, Pallikaris IG, Karantanas AH et al. High resolution MR eye protocol optimization: comparison between 3D-CISs, 3D-PSIF and 3D-VIBE sequences. Phys Med 2015; 10(15): 1120–1129. doi: 10.1016/ j.ejmp.2015.03.009.
30. Krohn-Hansen D, Zhang L, Haaskjold E, Meling TR, Nicolaissen B, Sjaastad I. Surgical anatomy of the superior orbit on ultra-high-resolution MRI at 9.4 Tesla. J Plast Surg Hand Surg 2015; 1 : 1–5.
31. Aletich V, Misra M, Shownkeen H, Camras LR, Debrun G. Evaluation and endovascular treatment of juxtaorbital vascular anomalies. Radiol Clin North Am 1999; 37(1): 123–133.
32. Hui KH, Pfeiffer ML, Esmaeli B. Value of positron emission tomography/ computed tomography in diagnosis and staging of primary ocular and orbital tumors. Saudi J Ophthalmol 2012; 26(4): 365–371. doi: 10.1016/ j.sjopt.2012.08.008.
33. Astrup J. Natural history and clinical management of optic pathway glioma. Br J Neurosurg 2003; 17(4): 327–335.
34. Egan RA, Lessell S. A contribution to the natural history of optic nerve sheath meningiomas. Arch Ophthalmol 2002; 120(11): 1505–1508.
35. Tow SL, Chandela S, Miller NR, Avellino AM.Long-term outcome in children with gliomas of the anterior visual pathway. Pediatr Neurol 2003; 28(4): 262–270.
36. Leonard JR, Perry A, Rubin JB, King AA, Chicoine MR, Gutmann DH. The role of surgical biopsy in the diagnosis of glioma in individuals with neurofibromatosis 1. Neurology 2006; 67(8): 1509–1512.
37. Bernardini FP, Bazzan M. Lymphoproliferative disease of the orbit. Curr Opin Ophthalmol 2007; 18(5): 398–401.
38. Leber KA, Berglöff J, Pendl G. Dose-response tolerance of the visual pathways and cranial nerves of the cavernous sinus to stereotactic radiosurgery. J Neurosurg 1998; 88(1): 43–50.
39. Tishler RB, Loeffler JS, Lunsford LD, Duma C, Alexander E jr, Kooy HM et al. Tolerance of cranial nerves of the cavernous sinus to radiosurgery. Int J Radiat Oncol Biol Phys 1993; 27(2): 215–221.
40. Stafford SL, Pollock BE, Leavitt JA, Foote RL, Brown PD, Link MJ et al. A study on the radiation tolerance of the optic nerves and chiasm after stereotactic radiosurgery. Int J Radiat Oncol Biol Phys 2003; 55(5): 1177–1181.
41. Mayo C, Martel MK, Marks LB, Flickinger J, Nam J, Kirkpatrick J. Radiation dose-volume effects of optic nerves and chiasm. Int J Radiat Oncol Biol Phys 2010; 76 (Suppl 3): S28–S35. doi: 10.1016/ j.ijrobp.2009.07.1753.
42. Eddleman CS, Liu JK. Optic nerve sheath meningioma: current diagnosis and treatment. Neurosurg Focus 2007; 23(5): E4.
43. Jo KI, Im YS, Kong DS, Seol HJ, Nam DH, Kim YD et al. Multisession Gamma Knife surgery for benign orbital tumors. J Neurosurg 2012; 117 (Suppl): 102–107. doi: 10.3171/ 2012.7.GKS12780.
44. Kortmann RD, Timmermann B, Taylor RE, Scarzello G, Plasswilm L, Paulsen F et al. Current and future strategies in radiotherapy of childhood low-grade glioma of the brain. Part II: treatment-related late toxicity. Strahlenther Onkol 2003; 179(9): 585–597.
45. Kortmann RD, Timmermann B, Taylor RE, Scarzello G, Plasswilm L, Paulsen F et al. Current and future strategies in radiotherapy of childhood low-grade glioma of the brain. Part I: treatment modalities of radiation therapy. Strahlenther Onkol 2003; 179(8): 509–520.
46. Landert M, Baumert BG, Bosch MM, Lutolf UM, Landau K. The visual impact of fractionated stereotactic conformal radiotherapy on seven eyes with optic nerve sheath meningiomas. J Neuroophthalmol 2005; 25(2): 86–91.
47. Liu D, Xu D, Zhang Z, Zhang Y, Li Y, Liu X et al. Long-term results of Gamma Knife surgery for optic nerve sheath meningioma. J Neurosurg 2010; 113 (Suppl): 28–33.
48. Richards JC, Roden D, Harper CS. Management of sight-threatening optic nerve sheath meningioma with fractionated stereotactic radiotherapy. Clin Experiment Ophthalmol 2005; 33(2): 137–141.
49. Smith JL, Vuksanovic MM, Yates BM, Bienfang DC. Radiation therapy for primary optic nerve meningiomas. J Clin Neuroophthalmol 1981; 1(2): 85–99.
50. Turbin RE, Pokorny K. Diagnosis and treatment of orbital optic nerve sheath meningioma. Cancer Control 2004; 11(5): 334–341.
51. Xu D, Liu D, Zhang Z, Zhang Y, Li Y, Liu X et al. Gamma Knife surgery in the management of orbital tumors. J Neurosurg 2010; 113 (Suppl): 34–38.
52. Cockerham KP, Bejjani GK, Kennerdell JS, Maroon JC. Surgery for orbital tumors. Part II: transorbital approaches. Neurosurg Focus 2001; 10(5): E3.
53. Bejjani GK, Cockerham KP, Kennerdel JS, Maroon JC. A reappraisal of surgery for orbital tumors. Part I: extraorbital approaches. Neurosurg Focus 2001; 10(5): E2.
54. Kennerdell JS, Maroon JC, Malton ML. Surgical approaches to orbital tumors. Clin Plast Surg 1988; 15(2): 273–282.
55. Maroon JC, Kennerdell JS. Surgical approaches to the orbit. Indications and techniques. J Neurosurg 1984; 60(6): 1226–1235.
56. Novák Z, Pábl L, Nádvorník P, Chrastina J. Stereotaktická biopsie nádoru očnice. Čes Slov Oftal 1997; 53(4): 220–222.
57. Berke RN. A modified Krönlein operation. Trans Am Ophthalmol Soc 1953; 51 : 193–231.
58. Galbraith JE, Sullivan JH. Decompression of the perioptic meninges for relief of papilledema. Am J Ophthalmol 1973; 76(5): 687–692.
59. Lipina R, Matoušek P, Paleček T, Komínek P. Rozšířený transnaazální endoskopický přístup k nádorům lebeční spodiny a očnice. Cesk Slov Neurol N 2012; 75/ 108(3): 326–332.
60. Matoušek P, Lipina R, Komínek P, Šmehlík P, Krejčí T, Syrovátka J et al. Transnazální endoskopická chirurgie nádorů očnice. Čes Slov Oftal 2012; 68(5): 202–206.
61. Netuka D, Masopust V, Belšán T, Profantová N, Be-neš V. Endoscopic endonasal resection of medial orbital lesions with intraoperative MRI. Acta Neurochir (Wien) 2013; 155(3): 455–461. doi: 10.1007/ s00701-012-1585-9.
62. Paluzzi A, Gardner PA, Fernandez-Miranda JC, Tormenti MJ, Stefko ST, Snyderman CH et al. „Round-the-Clock“ Surgical Access to the Orbit. J Neurol Surg B Skull Base 2015; 76(1): 12–24. doi: 10.1055/ s-0033-1360580.
63. Hassler WE, Meyer B, Rohde V, Unsold R. Pterional approach to the contralateral orbit. Neurosurgery 1994; 34(3): 552–524.
64. Raveh J, Laedrach K, Speiser M, Chen J, Vuillemin T, Seiler R et al. The subcranial approach for fronto-orbital and anteroposterior skull-base tumors. Arch Otolaryngol Head Neck Surg 1993; 119(4): 385–393.
65. Chynoranský M, Furdová A, Oláh Z. Exenterácie očnice. Čes Slov Oftal 1994; 50(2): 92–97.
66. Krásný J, Novák V, Otradovec J. Orbitální protéza po exenteraci očnice se zachováním víček a spojivkového vaku. Čes Slov Oftal 2006; 62(2): 94–99.
67. Rootman JCB. Orbital surgery: a conceptual approach. 2nd ed. Philadelphia: Lippincott, Williams and Wilkins 2014.
68. Baino F, Perero S, Ferraris S, Miola M, Balagna C, Verné E et al. Biomaterials for orbital implants and ocular prostheses: overview and future prospects. Acta Biomater 2014; 10(3): 1064–1087. doi: 10.1016/ j.actbio.2013.12.014.
69. Modugno A, Mantelli F, Sposato S, Moretti C, Lambiase A, Bonini S. Ocular prostheses in the last century: a retrospective analysis of 8,018 patients. Eye (Lond) 2013; 27(7): 865–870. doi: 10.1038/ eye.2013.97.
70. Gao D, Fei Z, Jiang X, Zhang X, Liu W, Fu L et al. The microsurgical treatment of cranio-orbital tumors assisted by intraoperative electrophysiologic monitoring and neuronavigation. Clin Neurol Neurosurg 2012; 114(7): 891–896. doi: 10.1016/ j.clineuro.2012.01.050.
71. Wilde F, Schramm A. Intraoperative imaging in orbital and midface reconstruction. Facial Plast Surg 2014; 30(5): 545–553. doi: 10.1055/ s-0034-1393700.
72. López JR. Neurophysiologic intraoperative monitoring of the oculomotor, trochlear, and abducens nerves. J Clin Neurophysiol 2011; 28(6): 543–550. doi: 10.1097/ WNP.0b013e31823da47e.
73. Dale EL, Strait TA, Sargent LA. Orbital reconstruction for pulsatile exophthalmos secondary to sphenoid wing dysplasia. Ann Plast Surg 2014; 72(6): S107–S111. doi: 10.1097/ SAP.0000000000000090.
74. Castillo BV jr, Kaufman L. Pediatric tumors of the eye and orbit. Pediatr Clin North Am 2003; 50(1): 149–172.
75. Bullock JD, Goldberg SH, Rakes SM. Orbital tumors in children. Ophthal Plast Reconstr Surg 1989; 5(1): 13–16.
76. Shields JA, Bakewell B, Augsburger JJ, Donoso LA, Bernardino V. Space-occupying orbital masses in children. A review of 250 consecutive biopsies. Ophthalmology 1986; 93(3): 379–384.
77. Shields JA, Shields CL, Scartozzi R. Survey of 1,264 patients with orbital tumors and simulating lesions: The 2002 Montgomery Lecture, part 1. Ophthalmology 2004; 111(5): 997–1008.
78. Shamji MF, Benoit BG. Syndromic and sporadic pediatric optic pathway gliomas: review of clinical and histopathological differences and treatment implications. Neurosurg Focus 2007; 23(5): E3.
79. Zoeller GK, Brathwaite CD, Sandberg DI. Malignant transformation of an optic pathway glioma without prior radiation therapy. J Neurosurg Pediatr 2010; 5(5): 507–510. doi: 10.3171/ 2009.12.PEDS09173.
80. Chutorian AM, Schwartz JF, Evans RA, Carter S. Optic gliomas in children. Neurology 1964; 14 : 83–95.
81. Russell DS, Rubinstein LJ. Pathology of tumours of the nervous system. 5th ed. Baltimore: Williams and Wilkins 1989.
82. Avery RA, Fisher MJ, Liu GT. Optic pathway gliomas. J Neuroophthalmol 2011; 31(3): 269–278. doi: 10.1097/ WNO.0b013e31822aef82.
83. Binning MJ, Liu JK, Kestle JR, Brockmeyer DL, Walker ML. Optic pathway gliomas: a review. Neurosurg Focus 2007; 23(5): E2.
84. Piccirilli M, Lenzi J, Delfinis C, Trasimeni G, Salvati M, Raco A. Spontaneous regression of optic pathways gliomas in three patients with neurofibromatosis type I and critical review of the literature. Childs Nerv Syst 2006; 22(10): 1332–1337.
85. Rozen WM, Joseph S, Lo PA. Spontaneous regression of low-grade gliomas in pediatric patients without neurofibromatosis. Pediatr Neurosurg 2008; 44(4): 324–328. doi: 10.1159/ 000134925.
86. Gajjar A, Bhargava R, Jenkins JJ, Heideman R, Sanford RA, Langston JW et al. Low-grade astrocytoma with neuraxis dissemination at diagnosis. J Neurosurg 1995; 83(1): 67–71.
87. Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis 1: controversies and recommendations. Ann Neurol 2007; 61(3): 189–198.
88. Hernáiz Driever P, von Hornstein S, Pietsch T, Kortmann R, Warmuth-Metz M, Emser A et al. Natural history and management of low-grade glioma in NF-1 children. J Neurooncol 2010; 100(2): 199–207. doi: 10.1007/ s11060-010-0159-z.
89. Otradovec J. Gliomy optiku chiasmatu (klinický obraz a diagnostika). Čs Oftal 1976; 32(1): 1–12.
90. Dodge HW jr, Love JG, Craig WM, Dockerty MB, Kearns TP, Holman CB et al. Gliomas of the optic nerves. AMA Arch Neurol Psychiatry 1958; 79(6): 607–621.
91. Taylor T, Jaspan T, Milano G, Gregson R, Parker T, Ritzmann T et al. Radiological classification of optic pathway gliomas: experience of a modified functional classification system. Br J Radiol 2008; 81(970): 761–766. doi: 10.1259/ bjr/ 65246351.
92. McCullough DC, Epstein F. Optic pathway tumors. A review with proposals for clinical staging. Cancer 1985; 56 (Suppl 7): 1789–1791.
93. Hollander MD, FitzPatrick M, O‘Connor SG, Flanders AE, Tartaglino LM. Optic gliomas. Radiol Clin North Am 1999; 37(1): 59–71.
94. Jost SC, Ackerman JW, Garbow JR, Manwaring LP, Gutmann DH, McKinstry RC. Diffusion-weighted and dynamic contrast-enhanced imaging as markers of clinical behavior in children with optic pathway glioma. Pediatr Radiol 2008; 38(12): 1293–1239. doi: 10.1007/ s00247-008-1003-x.
95. Weizman L, Ben Sira L, Joskowicz L, Constantini S, Precel R, Shofty B et al. Automatic segmentation, internal classification, and follow-up of optic pathway gliomas in MRI. Med Image Anal 2012; 16(1): 177–188. doi: 10.1016/ j.media.2011.07.001.
96. Shofty B, Weizman L, Joskowicz L, Constantini S, Kesler A, Ben-Bashat D et al. MRI internal segmentation of optic pathway gliomas: clinical implementation of a novel algorithm. Childs Nerv Syst 2011; 27(8): 1265–1272. doi: 10.1007/ s00381-011-1436-7.
97. Nicolin G, Parkin P, Mabbott D, Hargrave D, Bartels U, Tabori U et al. Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer 2009; 53(7): 1231–1237. doi: 10.1002/ pbc.22198.
98. Shofty B, Ben-Sira L, Freedman S, Yalon M, Dvir R, Weintraub M et al. Visual outcome following chemotherapy for progressive optic pathway gliomas. Pediatr Blood Cancer 2011; 57(3): 481–485. doi: 10.1002/ pbc.22-967.
99. Fouladi M, Wallace D, Langston JW, Mulhern R, Rose SR, Gajjar A et al. Survival and functional outcome of children with hypothalamic/ chiasmatic tumors. Cancer 2003; 97(4): 1084–1092.
100. Shofty B, Ben-Sira L, Kesler A, Constantini S. Optic pathway gliomas. In: Albright AL, Pollack IF, Adelson PD (eds). Principles and practice of pediatric neurosurgery. 3rd ed. New York: Thieme 2015.
101. Fisher BJ, Leighton CC, Vujovic O, Macdonald DR, Stitt L. Results of a policy of surveillance alone after surgical management of pediatric low grade gliomas. Int J Radiat Oncol Biol Phys 2001; 51(3): 704–710.
102. Beyer RA, Paden P, Sobel DF, Flynn FG. Moyamoya pattern of vascular occlusion after radiotherapy for glioma of the optic chiasm. Neurology 1986; 36(9): 1173–1178.
103. Sharif S, Ferner R, Birch JM, Gillespie JE, Gattamaneni HR, Baser ME et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol 2006; 24(16): 2570–2575.
104. Merchant TE, Hua CH, Shukla H, Ying X, Nill S, Oelfke U. Proton versus photon radiotherapy for common pediatric brain tumors: comparison of models of dose characteristics and their relationship to cognitive function. Pediatr Blood Cancer 2008; 51(1): 110–117. doi: 10.1002/ pbc.21530.
105. Kwon Y, Bae JS, Kim JM, Lee DH, Kim SY, Ahn JS et al. Visual changes after gamma knife surgery for optic nerve tumors. Report of three cases. J Neurosurg 2005; 102 (Suppl): 143–146.
106. Hug EB, Muenter MW, Archambeau JO, DeVries A, Liwnicz B, Loredo LN et al. Conformal proton radiation therapy for pediatric low-grade astrocytomas. Strahlenther Onkol 2002; 178(1): 10–17.
107. Ahn Y, Cho BK, Kim SK, Chung YN, Lee CS, Kim IH et al. Optic pathway glioma: outcome and prognostic factors in a surgical series. Childs Nerv Syst 2006; 22(9): 1136–1142.
108. Wright JE, Call NB, Liaricos S. Primary optic nerve meningioma. Br J Ophthalmol 1980; 64(8): 553–558.
109. Dutton JJ. Optic nerve sheath meningiomas. Surv Ophthalmol 1992; 37(3): 167–183.
110. Castel A, Boschi A, Renard L, De Potter P. Optic nerve sheath meningiomas: clinical features, functional prognosis and controversial treatment. Bull Soc Belge Ophtalmol 2000; 275 : 73–78.
111. Otradovec J, Votočková J. Primární meningeomy očnice (meningeomy optiku). Čs Oftal 1973; 29(3): 221–231.
112. Schick U, Dott U, Hassler W. Surgical management of meningiomas involving the optic nerve sheath. J Neurosurg 2004; 101(6): 951–959.
113. Misra S, Misra N, Gogri P, Mehta R. A rare case of bilateral optic nerve sheath meningioma. Indian J Ophthalmol 2014; 62(6): 728–730. doi: 10.4103/ 0301-4738.136238.
114. Trobe JD, Glaser JS, Post JD, Page LK. Bilateral optic canal meningiomas: a case report. Neurosurgery 1978; 3(1): 68–74.
115. Carrasco JR, Penne RB. Optic nerve sheath meningiomas and advanced treatment options. Curr Opin Ophthalmol 2004; 15(5): 406–410.
116. Kennerdell JS, Maroon JC, Malton M, Warren FA. The management of optic nerve sheath meningiomas. Am J Ophthalmol 1988; 106(4): 450–457.
117. Kim JW, Rizzo JF, Lessell S. Controversies in the management of optic nerve sheath meningiomas. Int Ophthalmol Clin 2005; 45(4): 15–23.
118. Wright JE, McNab AA, McDonald WI. Primary optic nerve sheath meningioma. Br J Ophthalmol 1989; 73(12): 960–966.
119. Killer HE, Laeng HR, Flammer J, Groscurth P. Architecture of arachnoid trabeculae, pillars, and septa in the subarachnoid space of the human optic nerve: anatomy and clinical considerations. Br J Ophthalmol 2003; 87(6): 777–781.
120. Sheikh BY, Siqueira E, Dayel F. Meningioma in children: a report of nine cases and a review of the literature. Surg Neurol 1996; 45(4): 328–435.
121. Liu JK, Forman S, Moorthy CR, Benzil DL. Update on treatment modalities for optic nerve sheath meningiomas. Neurosurg Focus 2003; 14(5): e7.
122. Sibony PA, Krauss HR, Kennerdell JS, Maroon JC, Slamovits TL. Optic nerve sheath meningiomas. Clinical manifestations. Ophthalmology 1984; 91(11): 1313–1326.
123. Mafee MF, Goodwin J, Dorodi S. Optic nerve sheath meningiomas. Role of MR imaging. Radiol Clin North Am 1999; 37(1): 37–58.
124. Lindblom B, Truwit CL, Hoyt WF. Optic nerve sheath meningioma. Definition of intraorbital, intracanalicular, and intracranial components with magnetic resonance imaging. Ophthalmology 1992; 99(4): 560–566.
125. Kanamalla US. The optic nerve tram-track sign. Radiology 2003; 227(3): 718–719.
126. Turbin RE, Thompson CR, Kennerdell JS, Cockerham KP, Kupersmith MJ. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology 2002; 109(5): 890–899.
127. Saeed P, Rootman J, Nugent RA, White VA, Mackenzie IR, Koornneef L. Optic nerve sheath meningiomas. Ophthalmology 2003; 110(10): 2019–2030.
128. Jeremic B, Pitz S. Primary optic nerve sheath meningioma: stereotactic fractionated radiation therapy as an emerging treatment of choice. Cancer 2007; 110(4): 714–722.
129. Fineman MS, Augsburger JJ. A new approach to an old problem. Surv Ophthalmol 1999; 43(6): 519–524.
130. Mark LE, Kennerdell JS, Maroon JC, Rosenbaum AE, Heinz R, Johnson BL. Microsurgical removal of a primary intraorbital meningioma. Am J Ophthalmol 1978; 86(5): 704–709.
131. Roser F, Nakamura M, Martini-Thomas R, Samii M, Tatagiba M. The role of surgery in meningiomas involving the optic nerve sheath. Clin Neurol Neurosurg 2006; 108(5): 470–476.
132. Delfini R, Missori P, Tarantino R, Ciapetta P, Cantore G. Primary benign tumors of the orbital cavity: comparative data in a series of patients with optic nerve glioma, sheath meningioma, or neurinoma. Surg Neurol 1996; 45(2): 147–153.
133. Ito M, Ishizawa A, Miyaoka M, Sato K, Ishii S. Intraorbital meningiomas. Surgical management and role of radiation therapy. Surg Neurol 1988; 29(6): 448–453.
134. Lee AG, Woo SY, Miller NR, Safran AB, Grant WH, Butler EB. Improvement in visual function in an eye with a presumed optic nerve sheath meningioma after treatment with three-dimensional conformal radiation therapy. J Neuroophthalmol 1996; 16(4): 247–251.
135. Cantore WA. Neural orbital tumors. Curr Opin Ophthalmol 2000; 11(5): 367–371.
136. Messmer EP, Camara J, Boniuk M, Font RL. Amputation neuroma of the orbit. Report of two cases and review of the literature. Ophthalmology 1984; 91(11): 1420–1423.
137. Carroll GS, Haik BG, Fleming JC, Weiss RA, Mafee MF. Peripheral nerve tumors of the orbit. Radiol Clin North Am 1999; 37(1): 195–202.
138. De Potter P, Shields CL, Shields JA, Rao VM, Eagle RC jr, Trachtenberg WM. The CT and MRI features of an unusual case of isolated orbital neurofibroma. Ophthal Plast Reconstr Surg 1992; 8(3): 221–227.
139. Garg R, Dhawan A, Gupta N, D‘souza P. A rare case of benign isolated schwannoma in the inferior orbit. Indian J Ophthalmol 2008; 56(6): 514–515.
140. Wang Y, Xiao LH. Orbital schwannomas: findings from magnetic resonance imaging in 62 cases. Eye (Lond) 2008; 22(8): 1034–1039.
141. Xian J, Zhang Z, Wang Z, Li J, Yang B, Chen Q et al. Evaluation of MR imaging findings differentiating cavernous haemangiomas from schwannomas in the orbit. Eur Radiol 2010; 20(9): 2221–2228. doi: 10.1007/ s00330-010-1774-y.
142. Cockerham KP, Cockerham GC, Stutzman R, Hidayat AA, Depper MH, Turbin RE et al. The clinical spectrum of schwannomas presenting with visual dysfunction: a clinico-pathologic study of three cases. Surv Ophthalmol 1999; 44(3): 226–234.
143. Khwarg SI, Lucarelli MJ, Lemke BN, Smith ME, Albert DM. Ancient schwannoma of the orbit. Arch Ophthalmol 1999; 117(2): 262–264.
144. Bhatnagar AK, Gerszten PC, Ozhasaglu C, Vogel WJ, Kalnicki S, Welch WC et al. CyberKnife frameless radiosurgery for the treatment of extracranial benign tumors. Technol Cancer Res Treat 2005; 4(5): 571–576.
145. Goh AS, Kim YD, Woo KI, Lee JI. Benign orbital apex tumors treated with multisession gamma knife radiosurgery. Ophthalmology 2013; 120(3): 635–641. doi: 10.1016/ j.ophtha.2012.08.015.
146. Hayashi M, Chernov M, Tamura N, Yomo S, Ochiai T, Nagai M et al. Gamma knife surgery for abducent nerve schwannoma. Report of 4 cases. J Neurosurg 2010; 113 (Suppl): 136–143.
147. Kim BS, Im YS, Woo KI, Kim YD, Lee JI. Multisession Gamma Knife Radiosurgery for Orbital Apex Tumors. World Neurosurg 2015; 84(4): 1005–1013. doi: 10.1016/ j.wneu.2015.04.042.
148. Kiratli H, Yildiz S, Soylemezoglu F. Neurofibromatosis type 2: optic nerve sheath meningioma in one orbit, intramuscular schwannoma in the other. Orbit 2008; 27(6): 451–454. doi: 10.1080/ 01676830802350356.
149. Aydin MD, Yildirim U, Gundogdu C, Dursun O, Uysal HH, Ozdikici M. Malignant peripheral nerve sheath tumor of the orbit: case report and literature review. Skull Base 2004; 14(2): 109–113.
150. Baran J, Falek A, Laskowski A, Sxeniuta A. Malignant schwannoma of the orbit in a 91-year old woman. Otolaryngol Pol 1995; 49 (Suppl 20): 494–497.
151. Erzurum SA, Melen O, Lissner G, Friedman DI, Sadun A, Feldon SE et al. Orbital malignant peripheral nerve sheath tumors. Treatment with surgical resection and radiation therapy. J Clin Neuroophthalmol 1993; 13(1): 1–7.
152. Fisher BJ, Dennis KE. Malignant epithelioid cranial nerve sheath tumor: case report of a radiation response. J Neurooncol 2006; 78(2): 173–177.
153. Chibbaro S, Herman P, Povlika M, George B. Malignant trigeminal schwannoma extending into the anterior skull base. Acta Neurochir (Wien) 2008; 150(6): 599–604. doi: 10.1007/ s00701-008-1563-4.
154. Miliaras G, Tsitsopoulos PP, Asproudis I, Tsekeris P, Polyzoidis K. Malignant orbital schwannoma with massive intracranial recurrence. Acta Neurochir (Wien) 2008; 150(12): 1291–1294. doi: 10.1007/ s00701-008-0159-3.
155. Romero-Rojas AE, Diaz-Pérez JA, Lozano-Castillo A. Malignant peripheric nerve sheath tumor of the orbit: first description of orbital location in a patient with NF1. Neurocirugia (Astur) 2010; 21(1): 37–45.
156. González-Orús Álvarez-Morujo R, García Leal R, Lasso Vázquez JM, Scola Yurrita B. Malignant peripheral nerve sheath tumour of the infra-orbital nerve. Neurocirugia (Astur) 2014; 25(5): 240–243. doi: 10.1016/ j.neucir.2014.02.002.
157. Lyons CJ, McNab AA, Garner A, Wright JE. Orbital malignant peripheral nerve sheath tumours. Br J Ophthalmol 1989; 73(9): 731–738.
158. D‘Agostino AN, Soule EH, Miller RH. Sarcomas of the peripheral nerves and somatic soft tissues associated with multiple neurofibromatosis (von Recklinghausen‘s disease). Cancer 1963; 16 : 1015–1027.
159. Wanebo JE, Malik JM, VandenBerg SR, Wanebo HJ, Driesen N, Persing JA. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 28 cases. Cancer 1993; 71(4): 1247–1253.
160. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986; 57(10): 2006–2021.
161. Aguiar Vitacca S, Sarrazin D, Henry-Amar M, Spielmann M, Genin J, Bernheim A et al. Neurosarcoma associated with Von Recklinghausen disease: apropos of 25 cases observed at the Gustave Roussy Institute from 1967 to 1990. Bull Cancer 1992; 79(1): 101–112.
162. Bonavolontà G, Strianese D, Grassi P, Comune C, Tranfa F, Uccello G et al. An analysis of 2,480 space-occupying lesions of the orbit from 1976 to 2011. Ophthal Plast Reconstr Surg 2013; 29(2): 79–86. doi: 10.1097/ IOP.0b013e31827a7622.
163. Char DH. Management of orbital tumors. Mayo Clin Proc 1993; 68(11): 1081–1096.
164. Lowe LH, Marchant TC, Rivard DC, Scherbel AJ. Vascular malformations: classification and terminology the radiologist needs to know. Semin Roentgenol 2012; 47(2): 106–117. doi: 10.1053/ j.ro.2011.11.002.
165. Yamasaki T, Handa H, Yamashita J, Paine JT, Tashiro Y, Uno A et al. Intracranial and orbital cavernous angiomas. A review of 30 cases. J Neurosurg 1986; 64(2): 197–208.
166. Sarraf D, Payne AM, Kitchen ND, Sehmi KS, Downes SM, Bird AC. Familial cavernous hemangioma: an expanding ocular spectrum. Arch Ophthalmol 2000; 118(7): 969–973.
167. Di Tommaso L, Scarpellini F, Salvi F, Ragazzini T, Foschini MP. Progesterone receptor expression in orbital cavernous hemangiomas. Virchows Arch 2000; 436(3): 284–288.
168. Henderson JW, Farrow GM, Garrity JA. Clinical course of an incompletely removed cavernous hemangioma of the orbit. Ophthalmology 1990; 97(5): 625–628.
169. Orcutt JC, Wulc AE, Mills RP, Smith CH. Asymptomatic orbital cavernous hemangiomas. Ophthalmology 1991; 98(8): 1257–1260.
170. Harris GJ, Jakobiec FA. Cavernous hemangioma of the orbit. J Neurosurg 1979; 51(2): 219–228.
171. Hejazi N, Classen R, Hassler W. Orbital and cerebral cavernomas: comparison of clinical, neuroimaging, and neuropathological features. Neurosurg Rev 1999; 22(1): 28–33.
172. Ansari SA, Mafee MF. Orbital cavernous hemangioma: role of imaging. Neuroimaging Clin N Am 2005; 15(1): 137–158.
173. Mafee MF, Putterman A, Valvassori GE, Campos M, Capek V. Orbital space-occupying lesions: role of computed tomography and magnetic resonance imaging. An analysis of 145 cases. Radiol Clin North Am 1987; 25(3): 529–559.
174. Scheuerle AF, Steiner HH, Kolling G, Kunze S, Aschoff A. Treatment and long-term outcome of patients with orbital cavernomas. Am J Ophthalmol 2004; 138(2): 237–244.
175. Zenobii M, Galzio RJ, Lucantoni D, Caffagni E, Magliani V. Spontaneous intraorbital hemorrhage caused by cavernous angioma of the orbit. J Neurosurg Sci 1984; 28(1): 37–40.
176. Khan AA, Niranjan A, Kano H, Kondziolka D, Flickinger JC, Lunsford LD. Stereotactic radiosurgery for cavernous sinus or orbital hemangiomas. Neurosurgery 2009; 65(5): 914–918. doi: 10.1227/ 01.NEU.0000356987.98197.71.
177. Liu X, Xu D, Zhang Y, Liu D, Song G. Gamma knife surgery in patients harboring orbital cavernous hemangiomas that were diagnosed on the basis of imaging findings. J Neurosurg 2010; 113 (Suppl): 39–43.
178. Haik BG, Karcioglu ZA, Gordon RA, Pechous BP. Capillary hemangioma (infantile periocular hemangioma). Surv Ophthalmol 1994; 38(5): 399–426.
179. Haywood RM, Monk BE, Mahaffey PJ. The treatment of early cutaneous capillary haemangiomata (strawberry naevi) with the tunable dye laser. Br J Plast Surg 2000; 53(4): 302–307.
180. Kushner BJ. The treatment of periorbital infantile hemangioma with intralesional corticosteroid. Plast Reconstr Surg 1985; 76(4): 517–526.
181. Gondová, G, Sejnová D, Hornová J, Elíz ová I, Gerinec A. Liečba kapilárního hemangiomu orbity betablokátormi. Folia Strabol Neuroophtalmol 2009; 10 (Suppl 1): 82–83.
182. Krásný J, Baráková D; Chodounský Z, Šach J. Lymfangiom orbitopalpebrální oblasti. Čes Slov Oftal 2014; 70(4): 152–159.
183. Iliff WJ, Green WR. Orbital lymphangiomas. Ophthalmology 1979; 86(5): 914–929.
184. Shields JA, Bakewell B, Augsburger JJ, Flanagan JC. Classification and incidence of space-occupying lesions of the orbit. A survey of 645 biopsies. Arch Ophthalmol 1984; 102(11): 1606–1611.
185. Volpe NJ, Jakobiec FA. Pediatric orbital tumors. Int Ophthalmol Clin 1992; 32(1): 201–221.
186. Vachharajani A, Paes B. Orbital lymphangioma with non-contiguous cerebral arteriovenous malformation, manifesting with thrombocytopenia (Kasabach-Merritt syndrome) and intracerebral hemorrhage. Acta Paediatr 2002; 91(1): 98–99.
187. Eiferman RA, Gushard RH. Chocolate cysts of the orbit. Ann Ophthalmol 1986; 18(4): 156–157.
188. Sires BS, Goins CR, Anderson RL, Holds JB. Systemic corticosteroid use in orbital lymphangioma. Ophthal Plast Reconstr Surg 2001; 17(2): 85–90.
189. Pacheco LF, Fernandes BF, Miyamoto C, Maloney SC, Arthurs B, Burnier MN jr. Rapid growth of an orbital hemangiopericytoma with atypical histopathological findings. Clin Ophthalmol 2014; 8 : 31–33. doi: 10.2147/ OPTH.S47901.
190. Billings KR, Fu YS, Calcaterra TC, Sercarz JA. Hemangiopericytoma of the head and neck. Am J Otolaryngol 2000; 21(4): 238–243.
191. Henderson JW, Farrow GM. Primary orbital hemangiopericytoma. An aggressive and potentiallymalignant neoplasm. Arch Ophthalmol 1978; 96(4): 666–673.
192. Jeeva I, Chang BY, Bagdonaite L, Kalantzis G, Fearnley TE, Ismail A et al. Treatment of orbital haemangiopericytoma with surgery and preoperative embolization. Eye (Lond) 2013; 27(2): 283–284. doi: 10.1038/ eye.2012.238.
193. Wallace KM, Alaraj A, Aakalu VK, Aletich V,Setabutr P. Endovascular preoperative embolization of orbital hemangiopericytoma with n-butyl cyanoacrylate glue. Ophthal Plast Reconstr Surg 2014; 30(4): e97–e100. doi: 10.1097/ IOP.0b013e3182a22bd8.
194. Gear HC, Kemp EG, Kacperek A, Errington RD. Treatment of recurrent orbital haemangiopericytoma with surgery and proton beam therapy. Br J Ophthalmol 2005; 89(1): 123–124.
195. Mafee MF, Pai E, Philip B. Rhabdomyosarcoma of the orbit. Evaluation with MR imaging and CT. Radiol Clin North Am 1998; 36(6): 1215–1227.
196. Crist WM, Anderson JR, Meza JL, Fryer C, Raney RB, Ruymann FB et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol 2001; 19(12): 3091–3102.
197. Shields JA, Shields CL. Rhabdomyosarcoma: review for the ophthalmologist. Surv Ophthalmol 2003; 48(1): 39–57.
198. Maurer HM, Beltangady M, Gehan EA, Crist W, Hammond D, Hays DM et al. The intergroup rhabdomyosarcoma study-I. A final report. Cancer 1988; 61(2): 209–220.
199. Abramson DH, Ellsworth RM, Tretter P, Wolff JA, Kitchin FD. The treatment of orbital rhabdomyosarcoma with irradiation and chemotherapy. Ophthalmology 1979; 86(7): 1330–1335.
200. Dalley RW. Fibrous histiocytoma and fibrous tissue tumors of the orbit. Radiol Clin North Am 1999; 37(1): 185–194.
201. Font RL, Hidayat AA. Fibrous histiocytoma of the orbit. A clinicopathologic study of 150 cases. Hum Pathol 1982; 13(3): 199–209.
202. Demirci H, Shields CL, Karatza EC, Shields JA. Orbital lymphoproliferative tumors: analysis of clinical features and systemic involvement in 160 cases. Ophthalmology 2008; 115(9): 1626–1631. doi: 10.1016/ j.ophtha.2008.02.004.
203. Demirci H, Shields CL, Shields JA, Honavar SG, Mercado GJ, Tovilla JC. Orbital tumors in the older adult population. Ophthalmology 2002; 109(2): 243–248.
204. Krásný J, Šach J, Brunnerová R, Konvička J, Jankovská M, Srp A et al. Orbitální tumory u dospělých – desetiletá studie. Čes Slov Oftal 2008; 64(6): 219–227.
205. Flanders AE, Espinosa GA, Markiewicz DA, Howell DD. Orbital lymphoma. Role of CT and MRI. Radiol Clin North Am 1987; 25(3): 601–613.
206. Vaněčková M, Seidl Z. Možnosti zobrazení očního bulbu, orbity a optického nervu v modalitě magnetické rezonance (MR). Část druhá: zobrazení očního nervu, orbity. Neurol Prax 2006; 3 : 164–167.
207. Sepahdari AR, Aakalu VK, Setabutr P, Shiehmorteza M, Naheedy JH, Mafee MF. Indeterminate orbital masses: restricted diffusion at MR imaging with echo-planar diffusion-weighted imaging predicts malignancy. Radiology 2010; 256(2): 554–564. doi: 10.1148/ radiol.10091956.
208. Vlková E, Kanova J. Maligní lymfom očnice. Čs Oftal 1986; 42(3): 196–200.
209. Marwaha G, Macklis R, Singh AD. Radiation therapy: orbital tumors. Dev Ophthalmol 2013; 52 : 94–101. doi: 10.1159/ 000351084.
210. Shields CL, Shields JA. Lacrimal gland tumors. Int Ophthalmol Clin 1993; 33(3): 181–188.
211. Jung WS, Ahn KJ, Park MR, Kim JY, Choi JJ, Kim BS et al. The radiological spectrum of orbital pathologies that involve the lacrimal gland and the lacrimal fossa. Korean J Radiol 2007; 8(4): 336–342.
212. Mafee MF, Edward DP, Koeller KK, Dorodi S. Lacrimal gland tumors and simulating lesions. Clinicopathologic and MR imaging features. Radiol Clin North Am 1999; 37(1): 219–239.
213. Wright JE, Stewart WB, Krohel GB. Clinical presentation and management of lacrimal gland tumours. Br J Ophthalmol 1979; 63(9): 600–606.
214. Stewart WB, Krohel GB, Wright JE. Lacrimal gland and fossa lesions: an approach to diagnosis and management. Ophthalmology 1979; 86(5): 886–895.
215. Bernardini FP, Devoto MH, Croxatto JO. Epithelial tumors of the lacrimal gland: an update. Curr Opin Ophthalmol 2008; 19(5): 409–413. doi: 10.1097/ ICU.0b013e32830b13e1.
216. Gamel JW, Font RL. Adenoid cystic carcinoma of the lacrimal gland: the clinical significance of a basaloid histologic pattern. Hum Pathol 1982; 13(3): 219–225.
217. Selva D, White VA, O‘Connell JX, Rootman J. Primary bone tumors of the orbit. Surv Ophthalmol 2004; 49(3): 328–342.
218. Yabut SM jr, Kenan S, Sissons HA, Lewis Mm. Malignant transformation of fibrous dysplasia. A case report and review of the literature. Clin Orthop Relat Res 1988; 228 : 281–289.
219. Edelstein C, Goldberg RA, Rubino G. Unilateral blindness after ipsilateral prophylactic transcranial optic canal decompression for fibrous dysplasia. Am J Ophthalmol 1998; 126(3): 469–471.
220. Katz BJ, Nerad JA. Ophthalmic manifestations of fibrous dysplasia: a disease of children and adults. Ophthalmology 1998; 105(12): 2207–2215.
221. Parisi MS, Oliveri MB, Mautalen CA. Bone mineral density response to long-term bisphosphonate therapy in fibrous dysplasia. J Clin Densitom 2001; 4(2): 167–172.
222. Lane JM, Khan SN, O‘Connor WJ, Nydick M, Hommen JP, Schneider R et al. Bisphosphonate therapy in fibrous dysplasia. Clin Orthop Relat Res 2001; 382 : 6–12.
223. Mewis-Levin L, Garcia CA, Olson JD. Plasma cell myeloma of the orbit. Ann Ophthalmol 1981; 13(4): 477–481.
224. Boari N, Gagliardi F, Spina A, Bailo M, Franzin A, Mortini P. Management of spheno-orbital en plaque meningiomas: clinical outcome in a consecutive series of 40 patients. Br J Neurosurg 2013; 27(1): 84–90. doi: 10.3109/ 02688697.2012.709557.
225. Oya S, Sade B, Lee JH. Sphenoorbital meningioma: surgical technique and outcome. J Neurosurg 2011; 114(5): 1241–1249. doi: 10.3171/ 2010.10.JNS101128.
226. Mirone G, Chibbaro S, Schiabello L, Tola S, George B. En plaque sphenoid wing meningiomas: recurrence factors and surgical strategy in a series of 71 patients. Neurosurgery 2009; 65 (Suppl 6): 100–109. doi: 10.1227/ 01.NEU.0000345652.19200.D5.
227. Cavazza S, Laffi GL, Lodi L, Gasparrini E, Tassinari G. Orbital dermoid cyst of childhood: clinical pathologic findings, classification and management. Int Ophthalmol 2011; 31(2): 93–97. doi: 10.1007/ s10792-011-9419-y.
228. Chung EM, Smirniotopoulos JG, Specht CS, Schroeder JW, Cube R. From the archives of the AFIP: pediatric orbit tumors and tumorlike lesions: nonosseous lesions of the extraocular orbit. Radiographics 2007; 27(6): 1777–1799.
229. Golden BA, Jaskolka MS, Ruiz RL. Craniofacial and orbital dermoids in children. Oral Maxillofac Surg Clin North Am 2012; 24(3): 417–425. doi: 10.1016/ j.coms.2012.04.006.
230. Günalp I, Gündüz K. Secondary orbital tumors. Ophthal Plast Reconstr Surg 1997; 13(1): 31–35.
231. Johnson TE, Tabbara KF, Weatherhead RG, Kersten RC, Rice C, Nasr AM. Secondary squamous cell carcinoma of the orbit. Arch Ophthalmol 1997; 115(1): 75–78.
232. Michel J, Fakhry N, Mancini J, Braustein D, Moreddu E, Giovanni A et al. Sinonasal squamous cell carcinomas: clinical outcomes and predictive factors. Int J Oral Maxillofac Surg 2014; 43(1): 1–6. doi: 10.1016/ j.ijom.2013.07.741.
233. Dulguerov P, Jacobsen MS, Allal AS, Leh-mann W, Calcaterra T. Nasal and paranasal sinus carcinoma: are we making progress? A series of 220 patients and a systematic review. Cancer 2001; 92(12): 3012–3029.
234. Imola MJ, Schramm VL jr. Orbital preservation in surgical management of sinonasal malignancy. Laryngoscope 2002; 112 : 1357–1365.
235. Lund VJ, Howard DJ, Wei WI, Cheesman AD. Craniofacial resection for tumors of the nasal cavity and paranasal sinuses – a 17-year experience. Head Neck 1998; 20(2): 97–105.
236. Iannetti G, Valentini V, Rinna C, Ventucci E, Marianetti TM. Ethmoido-orbital tumors: our experience. J Craniofac Surg 2005; 16(6): 1085–1091.
237. Patel SG, Singh B, Stambuk HE, Carlson D, Bridger PG, Cantu G et al. Craniofacial Surgery for Esthesioneuroblastoma: Report of an International Collaborative Study. J Neurol Surg B Skull Base 2012; 73(3): 208–220. doi: 10.1055/ s-0032-1311754.
238. Char DH, Miller T, Kroll S. Orbital metastases: diagnosis and course. Br J Ophthalmol 1997; 81(5): 386–390.
239. Goldberg RA, Rootman J, Cline RA. Tumors metastatic to the orbit: a changing picture. Surv Ophthalmol 1990; 35(1): 1–24.
240. Aggarwal SK, Bhavana K, Keshri A, Kumar R, Srivastava A. Frontal sinus mucocele with orbital complications: management by varied surgical approaches. Asian J Neurosurg. 2012; 7(3): 135–140. doi: 10.4103/ 1793-5482.103718.
241. Kuhn FA, Javer AR. Primary endoscopic management of the frontal sinus. Otolaryngol Clin North Am 2001; 34(1): 59–75.
242. Lešťák JV, Houšťava L, Pašta J, Voska P. Nádory orbity, muko-pyokély s propagací do očnice a jejich chirurgické řešení. Otorinolaryngol (Prague) 1996; 45(4): 177–181.
243. Antonelli V, Cremonini AM, Campobassi A, Pascarella R, Zofrea G, Servadei F. Traumatic encephalocele related to orbital roof fractures: report of six cases and literature review. Surg Neurol 2002; 57(2): 117–125.
244. Elster AD, Branch CL jr. Transalar sphenoidal encephaloceles: clinical and radiologic findings. Radiology 1989; 170(1): 245–247.
245. Morihara H, Zenke K, Shoda D, Fujiwara S, Suehiro S, Hatakeyama T. Intraorbital encephalocele in an adult patient presenting with pulsatile exophthalmos. Case report. Neurol Med Chir (Tokyo) 2010; 50(12):1126–1128.
246. Bahn RS. Graves’ ophthalmopathy. N Engl J Med2010; 362(8): 726–738. doi: 10.1056/ NEJMra0905750.
247. Fichter N, Guthoff RF, Schittkowski MP. Orbi-tal decompression in thyroid eye disease. ISRN Oph-thalmol 2012; 2012 : 739236. doi: 10.5402/ 2012/ 739236.
248. Boboridis KG, Uddin J, Mikropoulos DG, Bunce C,Mangouritsas G, Voudouragkaki IC et al. Critical Apprai-sal on Orbital Decompression for Thyroid Eye Dis-ease: a Systematic Review and Literature Search. Adv Ther 2015; 32(7): 595–611. doi: 10.1007/ s12325-015-0228-y
249. Rao R, MacIntosh PW, Yoon MK, Lefebvre DR. Current trends in the management of thyroid eye disease. Curr Opin Ophthalmol 2015; 26(6): 484–490. doi: 10.1097/ ICU.0000000000000203.
250. Boboridis KG, Bunce C. Surgical orbital decompression for thyroid eye disease. Cochrane Database Syst Rev 2011; 12: CD007630. doi: 10.1002/ 14651858.
251. Šlapák I. Chirurgická léčba primárních tumorů orbity. Otorinolaryng Foniat (Prague) 2008; 57(4): 206–209.
252. Klímová A, Diblík P, Kuthan P, Sklenka P. Zánětlivý pseudotumor očnice. Neurol Prax 2011; 12(3): 164–166.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2015 Issue 6
Most read in this issue
- Orbital Tumors
- European Society of Cardiology Congress
- Psychometric Properties of the Czech Version of Epworth Sleepiness Scale
- Familiar Amyloid Polyneuropathy – a Case Report