
Hereditární gelsolinová amyloidóza – klinické projevy a molekulárně genetická příčina
Hereditary gelsolin amyloidosis – clinical symptoms and molecular genetic cause
Aim: The aim of this study was to describe the clinical findings and molecular genetic cause of hereditary gelsolin amyloidosis in a family of Czech origin and a proband of Slovak origin. Patients and methods: Study participants underwent ophthalmic, neurological, nephrological, and cardiological examination. Sanger sequencing was used to screen the gelsolin gene (GSN). Results: Two mutations previously reported to be associated with hereditary gelsolin amyloidosis were identified; c.640G>T p. (Asp214Tyr) in a heterozygous state was found in seven individuals of Czech origin and c.640G>A p. (Asp214Asn) in the proband of Slovak origin. Linear corneal deposits were observed in the majority of affected subjects with the exception of two men aged 24 and 14 years. In addition to corneal deposits, patients in their fourth decade of life had drooping eyelids and carpal tunnel syndrome. Two oldest patients, aged 65 and 68 years, had also other typical signs of gelsolin amyloidosis, including dry eye syndrome, cutis laxa, and facial nerve lesion. The 68-year-old subject also had severe polyneuropathy, ataxia, dysarthria, and arrhythmia necessitating pacemaker implantation. Conclusion: Hereditary gelsolin amyloidosis should be included in the differential diagnosis of neuropathies and amyloidosis of unknown etiology. Since amyloid deposition in the cornea is easily detectable, ophthalmic examination has a crucial role in the diagnosis of this disease.
Keywords:
polyneuropathy – hereditary gelsolin amyloidosis – linear corneal deposits – facial nerve lesion
Authors:
P. Skalická 1,2; Ľ. Ďuďáková 2; A. Klímová 1; L. Huňa 1; C. J. Evans 3; M. Forgáč 4; O. Ulmanová 4; P. Mečíř 4; T. Paleček 5; V. Bednářová 6; V. Skovajsa 7; V. Skalníková 8; P. Lišková 1,2
Authors‘ workplace:
Oční klinika 1. LF UK a VFN v Praze
1; Laboratoř pro studium vzácných nemocí, Klinika pediatrie a dědičných poruch metabolismu 1. LF UK a VFN v Praze
2; UCL Institute of Ophthalmology, Londýn, Velká Británie
3; Neurologická klinika a Centrum klinických neurověd, 1. LF UK a VFN v Praze
4; II. interní klinika kardiologie a angiologie 1. LF UK a VFN v Praze
5; Klinika nefrologie 1. LF UK a VFN v Praze
6; Neurologie, Nemocnice Na Homolce, Praha
7; Kardiologie, Nemocnice Na Homolce, Praha
8
Published in:
Cesk Slov Neurol N 2021; 84(5): 449-455
Category:
Original Paper
doi:
https://doi.org/10.48095/cccsnn2021449
Overview
Cíl: Cílem této studie bylo popsat klinické nálezy a molekulárně genetickou příčinu hereditární gelsolinové amyloidózy v rodině českého původu a u probandky slovenského původu. Soubor a metody: Jedinci účastnící se studie podstoupili oční, neurologické, nefrologické a kardiologické vyšetření. Pomocí Sangerova sekvenování byl proveden screening genu gelsolin (GSN). Výsledky: Identifikovány byly dvě mutace, které byly již dříve popsány v souvislosti s hereditární gelsolinovou amyloidózou. Konkrétně u sedmi jedinců českého původu byla v heterozygotním stavu zjištěna c.640G>T p. (Asp214Tyr) a u probandky slovenského původu c.640G>A p. (Asp214Asn). U většiny nositelů patogenních variant byla přítomna lineární průhledná depozita v rohovkách, tato nebyla patrná pouze u dvou nejmladších mužů ve věku 24 a 14 let. U jedinců ve čtvrté dekádě života dominovaly jako klinické známky onemocnění kromě rohovkových depozit povislá oční víčka a syndrom karpálního tunelu. Dva nejstarší pacienti ve věku 65 a 68 let měli i další typické znaky gelsolinové amyloidózy: syndrom suchého oka, cutis laxa a parézu n. facialis. U 68letého muže byly dále zjištěny těžká polyneuropatie, ataxie, dysartrie a arytmie s nutností implantace kardiostimulátoru. Závěr: Hereditární gelsolinovou amyloidózu je třeba zahrnovat do diferenciální diagnózy neuropatií a amyloidóz neznámé etiologie. Vzhledem k tomu, že ukládání amyloidu v rohovkách je možné snadno detekovat, má oční vyšetření nezastupitelnou úlohu v diagnostickém procesu.
Klíčová slova:
hereditární gelsolinová amyloidóza – lineární depozita v rohovce – léze n. facialis – polyneuropatie
Úvod
Hereditární gelsolinová amyloidóza (HGA; Online Mendelian Inheritance in Man [OMIM] #105120) je vzácné pomalu progredující autozomálně dominantně dědičné onemocnění, které vzniká na podkladě akumulace mutovaného proteinu gelsolinu [1].
První symptomy HGA se obvykle objevují kolem 40. roku věku. Pacienti nejčastěji udávají nespecifické oční obtíže jako např. pocit suchého oka, podrážděnost a citlivost na světlo [2]. Nejvýznamnějšími klinickými znaky jsou pomalu progredující kraniální neuropatie, cutis laxa a lineární depozita v rohovce. V počátečních fázích může být HGA zaměněna za mřížkovou dystrofii rohovky typu 1 (OMIM #122200), která je podmíněna mutacemi v genu pro beta-indukovaný transformující růstový faktor (TGFBI) a je také charakterizována ukládáním lineárních depozit v rohovkách, na rozdíl od HGA ale nemá systémové projevy [3,4]. Kromě kraniální neuropatie (na první pohled mnohdy upoutá léze n. facialis) mohou u HGA vzniknout i poruchy CNS, periferních a autonomních nervů [5]. U některých pacientů jsou přítomny poruchy srdečního rytmu, kardiomyopatie i nefropatie. Popsána byla také těžká ataxie s fatálními následky [6]. Kožní problémy začínají poklesem kůže očních víček, posléze dochází k uvolnění kůže celého obličeje spolu se ztrátou mimiky a vrásek, což bývá popisováno jako „tvář podobající se masce“ [2]. Vyskytnout se může ale i řada jiných patologických nálezů jako např. glaukom, suchá kůže nebo abnormální tvorba modřin vznikající nejspíše na podkladě amyloidové angiopatie a trombocytopatie [2,7,8].
Gelsolin se exprimuje ve většině savčích buněk a jeho extracelulární izoforma je dále přítomna i v plazmě a mozkomíšním moku [9]. Bylo zjištěno, že tento protein má úlohu při modulaci aktinu a v apoptotickém procesu [10,11]. Přítomnost patogenní mutace v genu GSN vytváří proteolytické místo pro alfa-gelsolinázu, což vede ke vzniku 71 aminokyselin dlouhému fragmentu, který se ukládá v tkáních ve formě vláken amyloidu. V souladu s definicí této substance se barví Kongo červení a vykazuje zelený (apple-green) dvojlom v polarizovaném světle [12,13].
Celosvětově byly u pacientů s HGA zjištěny pouze tři patogenní mutace v GSN, přičemž u naprosté většiny je přítomna c.640G>A p. (Asp214Asn). Tato mutace je díky fenoménu zakladatele častá především ve Finsku. Odhaduje se, že zde žije 400–1 000 nositelů dané mutace, což v této populaci představuje prevalenci HGA jeden postižený na 5 500–13 750 obyvatel [2]. Kromě Finska byla tato patogenní varianta dále zjištěna v několika dalších populacích převážně evropského původu [12,14–23]. Druhá mutace c.640G>T p. (Asp214Tyr) vedoucí ke vzniku HGA je mnohem vzácnější, dosud byla identifikována pouze v pěti rodinách, a to českého, dánského, korejského, francouzského a brazilského původu [12,24–27]. Zcela nedávno byla v GSN nalezena nová pravděpodobně patogenní varianta c.1631T>G p. (Met544Arg) asociovaná s HGA [28].
Cílem této práce bylo popsat klinické nálezy a určit molekulárně genetickou příčinu ve dvou rodinách s HGA.
Metody
Do studie byli zařazeni příslušníci dvou rodin; rodina A byla slovenského původu, rodina B udávala původ český.
Molekulárně-genetické vyšetření
Extrakce DNA byla provedena z leukocytů venózní krve pomocí kitu Gentra Puregene Blood Kit (Qiagen, Hilden, Německo). Genomová DNA byla amplifikována pomocí námi navržených primerů pro exon 4 genu GSN (dostupné u autorů na dotaz) a dříve publikovaných primerů pro gen TGFBI [4]. Provedli jsme Sangerovo sekvenování získaných PCR produktů na kapilárním sekvenátoru ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Jako referenční sekvence byla použita NM_000177.4 (GSN) a NM_000358.2 (TGFBI).
Klinické vyšetření
Dostupní rodinní příslušníci byli podrobeni detailnímu očnímu vyšetření, vč. fotografie předního segmentu oka, vyšetření fundu v mydriáze a vyšetření makul pomocí optické koherenční tomografie se spektrální doménou (SD-OCT, Spectralis Heidelberg Engineering, Heidelberg, Německo). Zraková ostrost byla stanovena pomocí Snellenových optotypů a převedena na decimální hodnoty. Dále vyšetřovaní jedinci podstoupili základní biochemické vyšetření krve vč. ukazatelů renálních funkcí. Provedeny byly i sonografické vyšetření ledvin, analýza moči k vyloučení proteinurie a vyhodnocení vylučovací funkce ledvin. Neurologické vyšetření zahrnovalo EMG, EEG a kmenové sluchové evokované potenciály (brainstem auditory evoked potential; BAEP). Jeden pacient podstoupil také MR mozku, biopsii n. suralis a vyšetření likvoru. Komplexní neinvazivní kardiologické vyšetření zahrnovalo transtorakální echokardiografii a elektrokardiografii (EKG).
Výsledky
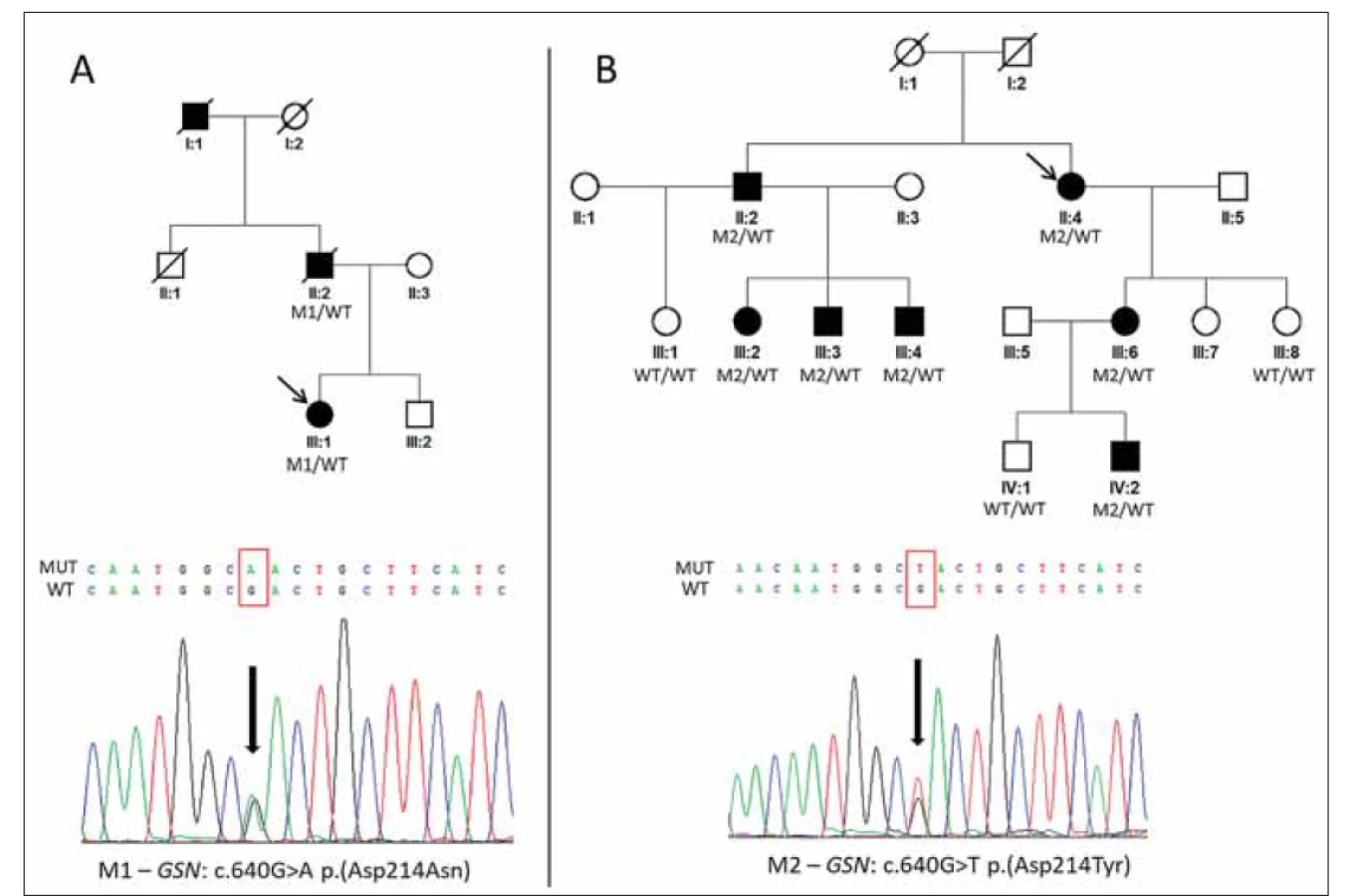
Probandka slovenského původu z rodiny A (III: 1; obr. 1A) byla odeslána na Oční kliniku 1. LF UK a VFN v Praze pro opakované záněty rohovky nejasné etiologie, jež byly posléze vyhodnoceny jako součást klinického obrazu dystrofie rohovek, pro kterou byla dlouhodobě sledována. Na základě zprávy z molekulárně genetického vyšetření otce, provedeného v minulosti v zahraniční laboratoři, byl indikován cílený screening exonu 4 genu GSN. Přítomnost mutace p. (Asp214Asn) v heterozygotním stavu potvrdila diagnózu HGA (obr. 1A).
(A) Rodina slovenského původu s výskytem heterozygotní mutace M1 c.640G>A p.(Asp214Asn) v genu GSN a její sekvenogram.
(B) Rodina českého původu s výskytem heterozygotní mutace M2 c.640G>T p.(Asp214Tyr) v genu GSN a její sekvenogram.
Probandky jsou v jednotlivých rodinách označeny šipkami.
GSN – gen gelsolin; M – mutace; WT – divoká alela
Fig. 1. Pedigrees of two families with hereditary gelsolin amyloidosis and detected causal mutations.
(A) Family of Slovak origin with identified heterozygous mutation M1 c.640G>A p.(Asp214Asn) in the GSN gene and its sequence
chromatogram.
(B) Family of Czech origin with identified heterozygous mutation M2 c.640G>T p.(Asp214Tyr) in the GSN gene and its sequence
chromatogram. Probands are indicated by arrows.
GSN – gelsolin gene; M – mutation; WT – wild type
Při očním vyšetření provedeném ve věku 40 let byla zdokumentována depozita v rohovkách a známky recidivujících erozí (tab. 1). Ztráta elasticity kůže v oblasti horních a dolních víček byla patrná z fotografie pořízené před jejich plastickou operací, kterou pacientka podstoupila ve 39 letech. Ostatní oční nálezy, vč. vyšetření sítnice v mydriáze a pomocí SD-OCT, byly zcela v normě. Neurologické a nefrologické vyšetření bylo také bez patologie (tab. 1). Echokardiograficky nebyly zjištěny známky suspektní z amyloidové infiltrace myokardu, jako vedlejší nález byla nalezena středně významná insuficience mitrální chlopně při prolapsu lehce ztluštělého předního cípu.
![Projevy hereditární gelsolinové amyloidózy u osmi jedinců pozitivně testovaných na přítomnost patogenní mutace v genu GSN.
Projevy onemocnění jsou porovnány s finskou kohortou 227 jedinců [2]. Jednici jsou označeni dle obr. 1 (A rodina slovenského původu,
B rodina českého původu) a seřazeni dle klesajícího věku. Údaje o postižení sluchu byly získány pouze anamnesticky, audiogram nebyl
proveden.](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/f962faf184a125055f24319b8d40fa9a.png)
Dle dostupné zdravotnické dokumentace měli otec (II: 1; obr. 1A) a dědeček (I: 1; obr. 1A) také dystrofii rohovek s opakovanými erozemi, u otce byl prokázán amyloid i v bioptickém vzorku ledvin.
Probandka z rodiny B (II: 4; obr. 1B) byla odeslána ke konziliárnímu vyšetření s diagnózou mřížkové dystrofie rohovky. Na základě nálezu lineárních rohovkových depozit (obr. 2G) byl indikován screening genu TGFBI, který ale nevedl k detekci příčinných mutací, proto byl posléze proveden screening exonu 4 genu GSN. Byla zjištěna již dříve popsaná patogenní mutace v heterozygotním stavu p. (Asp214Tyr) vedoucí ke vzniku HGA (obr. 1B). Sledováním v rodině byla potvrzena přítomnost této varianty celkem u šesti dalších rodinných příslušníků, u třech příbuzných pak byla vyloučena (obr. 1B). Výsledky všech jejich klinických vyšetření jsou přehledně zpracovány v tab. 1.
(A) Maskovitý vzhled obličeje 68letého muže (jedinec II:2) s ochablou kůží, povislými víčky a pokleslými koutky úst jako projev zjevné léze
n. facialis, která je obzvláště patrná na pravé straně.
(B) 65letá žena (II:4) s ochablými očními víčky, pokleslými koutky úst a mírně odstávajícím dolním rtem, zejména vlevo.
(C) 46letá žena (III:6) s povislou kůži v obličeji.
(D) 14letý nositel patogenní mutace (IV:2) bez klinických projevů hereditární gelsolinové amyloidózy.
(E) Lagoftalmus (šipky) u 68letého muže (II:2).
(F) Rohovka stejného pacienta (II:2) s lineárními depozity (šipka).
(G) Rohovka ženy (II:4) zobrazující lineární a větvící se depozita (šipka).
(H) Rohovka ženy (III:6) zobrazující lineární a větvící se depozita (šipka).
(I) Čirá rohovka asymptomatického muže (IV:2).
Fig. 2. Clinical findings in subject with hereditary gelsolin amyloidosis from family B.
(A) A 68-year-old male (II:2) with a mask-like facial appearance, cutis laxa, drooping eyelids and mouth as a result of obvious facial nerve
lesion, especially on the right side.
(B) A 65-year-old female (II:4) with drooping eyelids, mouth corners and mild protrusion of her lower lip, which is more pronounced on the
left side.
(C) A 46-year-old female (III:6) with drooping facial skin.
(D) A 14-year-old male with pathogenic mutation (IV:2) without clinical signs of hereditary gelsolin amyloidosis.
(E) Lagophthalmos (arrows) in a 68-year-old male (II:2).
(F) Cornea of the same patient (II:2) containing linear deposits (arrow).
(G) Cornea of female (II:4) showing linear branching deposits (arrow).
(H) Cornea of female (III:6) showing linear branching deposits (arrow).
(I) Clear cornea of asymptomatic male (IV:2).
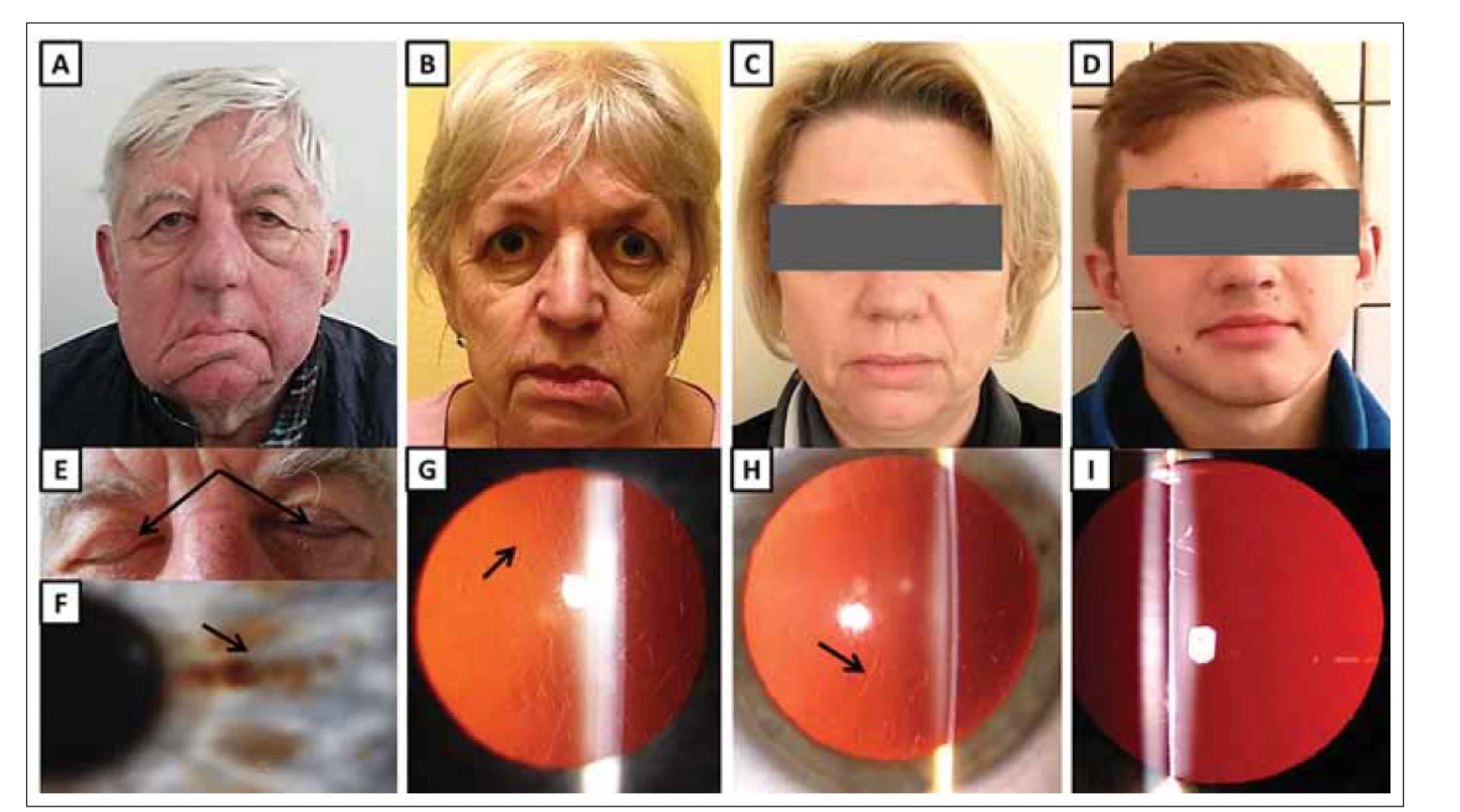
Probandka při prvním vyšetření ve věku 58 let udávala pět let trvající obtíže ve smyslu řezání a světloplachosti. Ve věku 65 let jsme u ní zdokumentovali bilaterálně ochablá oční víčka, lineární depozita ve stromatu rohovek a recidivující eroze rohovek (obr. 2B a G). V obličeji byly zaznamenány další charakteristické známky choroby: cutis laxa, pokleslé koutky úst a odstávající dolní ret na podkladě léze n. facialis, zvláště vlevo (obr. 2B).
Vyšetření EMG provedené též ve věku 65 let prokázalo oboustranně fokální demyelinizační lézi n. medianus v oblasti karpálního tunelu. Klinické neurologické vyšetření dále zachytilo pallanestezie, nicméně bez známek polyneuropatie při vyšetření EMG. Komplexní kardiologické vyšetření, zahrnující kromě echokardiografického a klidového vyšetření EKG též Holterovskou monitoraci EKG, nenalezlo změny svědčící pro postižení srdce.
U dcery probandky (III: 6; obr. 1B) jsme ve věku 46 let zaznamenali typická lineární depozita ve stromatu rohovek obou očí a povislou kůži v obličeji, zejména v oblasti očních víček (obr. 2C a H). Při kardiologickém, nefrologickém a klinickém neurologickém vyšetření nebyly zjištěny žádné významné abnormality. Pacientka však ve věku 45 let prodělala oboustrannou operaci karpálních tunelů.
Muž (IV: 1; obr. 1B), který byl negativně testován na přítomnost patogenní mutace, nevykazoval ve věku 25 let žádnou oční patologii. U 14letého jedince (IV: 2; obr. 1B), prokázaného nositele mutace p. (Asp214Tyr) také nebyla zjištěna depozita v rohovce (obr. 2D a I).
Bratr probandky z rodiny B (II: 2; obr. 1B) byl od 61 let sledován pro etiologicky nejasnou kraniální neuropatii manifestující se iniciálně jako pokleslý ústní koutek. Anamnesticky se dále zjistilo, že zhruba od tohoto věku trpí nejistotou při chůzi a špatnou výslovností, ke kterým se přibližně ve věku 65 let přidružila i porucha citlivosti prstů na noze. Opakované neurologické vyšetření vč. EMG svědčilo pro postupně progredující distální periferní motoricko-senzitivní neuropatii na končetinách a kraniální neuropatii. Na všech etážích, na končetinách i v oblasti kraniální (především v oblasti n. facialis a n. hypoglossus), byly jehlovým vyšetřením zjištěny známky chronické axonální denervace a kolaterální reinervace. Vyšetření MR, provedené ve věku 63 let, pouze s nálezem difuzní atrofie mozku, neozřejmilo etiologii obtíží. Rozsáhlý laboratorní screening zaměřený na neuropatie prokázal jen lehkou poruchu glukózové tolerance, ostatní provedená vyšetření vč. analýzy mozkomíšního moku byla v normě. Biopsie n. suralis potvrdila pokročilou axonální neuropatii s mírnou demyelinizační složkou a současně byla zachycena drobná depozita eosinofilního materiálu svědčící pro amyloidózu.
Pacient byl od 62 let v péči kardiologa pro paroxyzmální fibrilaci síní bez nutnosti antikoagulační terapie či chirurgické intervence a v 68 letech mu byl pro symptomatický sick sinus syndrom (stav po kolapsu) implantován kardiostimulátor. Tento nález však nebyl hodnocen jako jednoznačně související s HGA, neboť jak fibrilace síní, tak převodní porucha vedoucí k implantaci kardiostimulátoru jsou v tomto věku relativně časté. Ve věku 68 let byl u pacienta zjištěn a léčen karcinom prostaty bez generalizace.
Z oční patologie byla přítomna lineární průhledná depozita ve stromatu obou rohovek (tab. 1, obr. 2F). Fotografie pořízená v jeho 68 letech dokumentuje maskovitý vzhled obličeje s převislými víčky a pokleslými koutky úst (obr. 2A). Dalším klinickým projevem léze n. facialis je neschopnost uzavřít oční štěrbinu, tedy lagoftalmus (obr. 2E).
U 40leté ženy (III: 2; obr. 1B) byla lineární depozita v rohovkách zjištěna náhodně při preventivním vyšetření. Anamnesticky udávala od svých 38 let noční parestezie všech prstů obou horních končetin a od 40 let pociťovala brnění a bolesti v zápěstí i přes den. Pomocí EMG byla prokázána lehká oboustranná fokální demyelinizační léze n. medianus (tj. syndrom karpálního tunelu lehkého stupně). Pacientka je také pravidelně sledovaná pro karcinoid, který byl odhalen při appendektomii v jejích 37 letech.
Asymptomatický 37letý bratr (III: 3; obr. 1B), také prokázaný nositel patogenní mutace v genu GSN, měl zjištěna pouze rohovková depozita (tab. 1). U jeho 24letého bratra (III: 4; obr. 1B), také pozitivně testovaného na přítomnost mutace GSN, byla rohovka zatím čirá.
Diskuze
V této práci popisujeme klinické a molekulárně genetické nálezy u příslušníků jedné rodiny českého a jedné rodiny slovenského původu se vzácným onemocněním HGA. Přestože ve světě se jedná o známou klinickou jednotku, nebylo na ni u členů české rodiny v diagnostickém procesu dlouho pomýšleno.
Mutace c.640G>T v genu GSN zjištěná v české rodině byla dosud, vč. této studie, celosvětově popsána pouze v šesti rodinách. Vzhledem k tomu, že dvě z těchto rodin jsou českého původu, je možné, že mají společného předka. Tuto hypotézu bohužel nebylo možné ověřit, neboť příslušníky rodiny, jejichž nálezy byly publikovány v roce 1959 a 1992, se nepodařilo dohledat [12,24].
Přestože jsme vyšetřili pouze osm prokázaných nositelů patogenních mutací v genu GSN, lze říci, že jejich klinické nálezy odpovídaly popisu v literatuře a odrážely pomalu progredující charakter onemocnění. Jak bylo zjištěno studiem 227 finských pacientů s HGA, první známkou onemocnění, kterou jsme pozorovali i u našich pacientů, bývá přítomnost průhledných depozit v rohovkách [2]. Záchyt těchto změn je obvykle náhodný v průběhu očního vyšetření pro jiné, často nespecifické obtíže nebo při předpisu presbyopické brýlové korekce kolem 40. roku věku. Uloženiny, jež jsou typicky lineární a s věkem postupně přibývají od periferie do centra rohovky, nejsou příčinou významného poklesu zrakové ostrosti. Ten je nejpravděpodobněji zapříčiněn těžkou keratopatií a vznikem neurotrofických rohovkových ulcerací. Na rozdíl od finské kohorty jsme tyto komplikace nezaznamenali a ani jsme nebyli nuceni rohovky transplantovat [29]. Pouze u dvou našich jedinců jsme zjistili malý pokles vidění v důsledku nepravidelného astigmatismu bez prokazatelné souvislosti s HGA, konkrétně u 40leté ženy z rodiny A (III: 1) a u 65leté probandky z rodiny B (II: 4).
Závažnější projevy onemocnění jako léze n. facialis, polyneuropatie a arytmie se objevují víceméně současně v šestém decéniu [2]. Srdeční příznaky jsou méně časté; pouze u devíti finských pacientů (4 %) bylo nutné implantovat kardiostimulátor a 12 (6,1 %) mělo kardiomyopatii, což je však stále mnohem více v porovnání s běžnou populací [2,30,31]. V našem souboru měl nejzávažnější nálezy nejstarší 68letý jedinec z rodiny B (II: 2), u kterého se projevila těžká polyneuropatie zasahující jak kraniální nervy, tak i motorické a autonomní nervy a kterému jako jedinému byl implantován kardiostimulátor. U tohoto konkrétního pacienta je obtížné spojovat postižení srdce s HGA, protože mu nebyla provedena biopsie myokardu. Jeho celkový stav bohužel nedovoloval další invazivní vyšetření. U všech tří žen z rodiny B starších 40 let byl přítomen syndrom karpálního tunelu, který se vyskytuje u pacientů s HGA ve více než 35 % a může být i iniciální manifestací onemocnění [2]. Příčinou je progresivní infiltrace amyloidových vláken do ligamentum carpi a synoviální tkáně vedoucí ke kompresi n. medianus.
Onemocnění HGA není v současné době léčitelné, nicméně znalost základní diagnózy umožňuje cílená preventivní opatření. Vzhledem k riziku vzniku poruch srdečního rytmu, a tím i ohrožení náhlou smrtí, se domníváme, že by pacienti s HGA měli být po 50. roce věku dispenzarizováni, což dokládá i náhle vzniklý kolaps u nejstaršího pacienta z našeho souboru. Pravidelné sledování je dále důležité z hlediska možného rozvoje glaukomu. Pacienti by měli být také poučeni o snížené citlivosti rohovky, což zvyšuje pravděpodobnost vzniku komplikací, nejčastěji vředu rohovky, např. při drobném poranění [2,29].
Přestože může HGA u starších jedinců významně ovlivnit kvalitu jejich života, studium dokumentace 272 zemřelých finských pacientů ukázalo, že choroba nevede ke zkrácení průměrné délky života. V tomto souboru byla amyloidóza vyhodnocena jako základní příčina úmrtí u 19,9 % pacientů a autoři současně upozornili na zvýšené riziko fatálních renálních komplikací, přičemž malignity se vyskytovaly v porovnání s běžnou populací méně často. Proč dochází k tomuto jevu, nebylo dosud uspokojivě vysvětleno [32]. Amyloidóza ledvin u nositelů mutace p. (Asp214Asn) v homozygotním stavu může způsobit nefrotický syndrom již kolem 20. roku věku [33,34] a může být podceňována u heterozygotů [2], proto je třeba vždy pomýšlet na renální komplikace HGA. V naší skupině osmi nositelů patogenních mutací v genu GSN nebylo poškození ledvin prokázáno.
Postižení nervů a tkání vzniká u HGA ukládáním amyloidu. Tento typ amyloidózy lze diagnostikovat buď genetickým testováním nebo průkazem depozit amyloidu přímo v postižených tkáních či z biopsie nervů. Vzhledem k tomu, že u HGA byl dosud záchyt patogenní mutace stoprocentní, jeví se stanovení diagnózy na úrovni genu jako jednodušší, pacienta méně zatěžující.
Naše práce dokládá, že HGA by měla být i v České a Slovenské republice zahrnuta do diferenciální diagnostiky nejasných polyneuropatií, zvláště u případů s postižením kraniálních nervů. Jelikož je ukládání amyloidu v rohovkách snadno detekovatelné, má oční vyšetření svoji nezastupitelnou úlohu. Oftalmolog může být tedy první, který na toto dědičné onemocnění upozorní a zahájí molekulárně genetickou analýzu. Včasné určení správné diagnózy může pomoci předejít pozdějším závažným systémovým komplikacím HGA.
Etické aspekty
Práce byla provedena ve shodě s Helsinskou deklarací z roku 1975 a jejími revizemi v letech 2004 a 2008. Výzkum popsaný v této studii proběhl v souladu s doporučením Etické komise VFN (referenční číslo 55/18) a všichni účastníci nebo jejich zákonní zástupci podepsali informovaný souhlas. Všichni prezentovaní členové rodiny B souhlasí s uveřejněním fotografie obličeje, konkrétně muž II: 2 a žena II: 4 souhlasí se zobrazením celého obličeje, ostatní členové si přáli zakrýt oči. Autoři prohlašují, že s podobou obrázku byli všichni vyšetření jedinci a jejich zákonní zástupci seznámeni. Všichni zobrazení jedinci souhlasili se zveřejněním fotografií obličeje v prezentovaném formátu.
Poděkovaní
Děkujeme prof. MUDr. Josefu Zámečníkovi, Ph.D., za laskavou konzultaci nálezu svalové biopsie.
Grantová podpora
Práce byla podpořena grantem Ministerstva zdravotnictví ČR: AZV 17-32318A. Institucionální podpora Karlovy Univerzity: Progres Q27, Q26/LF1, UNCE/MED/007, SVV 260516. Veškerá práva podle předpisů na ochranu duševního vlastnictví jsou vyhrazena.
Konflikt zájmů
Autoři deklarují, že v souvislosti s předmětem studie nemají žádný konflikt zájmů.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
doc. MUDr. Petra Lišková, Ph.D.
Klinika pediatrie a dědičných poruch
metabolismu a Oční klinika
1. LF UK a VFN v Praze
Ke Karlovu 2
128 00 Praha
e-mail: petra.liskova@lf1.cuni.cz
Přijato k recenzi: 16. 5. 2021
Přijato do tisku: 16. 9. 2021
Sources
1. Maury CP. Homozygous familial amyloidosis, Finnish type: demonstration of glomerular gelsolin-derived amyloid and non-amyloid tubular gelsolin. Clin Nephrol 1993; 40 (1): 53–56.
2. Nikoskinen T, Schmidt EK, Strbian D et al. Natural course of Finnish gelsolin amyloidosis. Ann Med 2015; 47 (6): 506–511. doi: 10.3109/07853890.2015.1075 063.
3. Liskova P, Klintworth GK, Bowling BL et al. Phenotype associated with the H626P mutation and other changes in the TGFBI gene in Czech families. Ophthalmic Res 2008; 40 (2): 105–108. doi: 10.1159/000115325.
4. Evans CJ, Davidson AE, Carnt N et al. Genotype-phenotype correlation for TGFBI corneal dystrophies identifies p. (G623D) as a novel cause of epithelial basement membrane dystrophy. Invest Ophthalmol Vis Sci 2016; 57 (13): 5407–5414. doi: 10.1167/iovs.16-19818.
5. Makioka K, Ikeda M, Ikeda Y et al. Familial amyloid polyneuropathy (Finnish type) presenting multiple cranial nerve deficits with carpal tunnel syndrome and orthostatic hypotension. Neurol Res 2010; 32 (5): 472–475. doi: 10.1179/174313209X409007.
6. Tanskanen M, Paetau A, Salonen O et al. Severe ataxia with neuropathy in hereditary gelsolin amyloidosis: a case report. Amyloid 2007; 14 (1): 89–95. doi: 10.1080/13506120601116393.
7. Kiuru S, Salonen O, Haltia M. Gelsolin-related spinal and cerebral amyloid angiopathy. Ann Neurol 1999; 45 (3): 305–311. doi: 10.1002/1531-8249 (199903) 45 : 3<305:: aid-ana5>3.0.co; 2-e.
8. Kiuru S, Javela K, Somer H et al. Altered platelet shape change in hereditary gelsolin Asp187Asn-related amyloidosis. Thromb Haemost 2000; 83 (3): 491–495.
9. Bucki R, Kulakowska A, Byfield FJ et al. Plasma gelsolin modulates cellular response to sphingosine 1-phosphate. Am J Physiol Cell Physiol 2010; 299 (6): C1516–1523. doi: 10.1152/ajpcell.00051.2010.
10. Sun HQ, Yamamoto M, Mejillano M et al. Gelsolin, a multifunctional actin regulatory protein. J Biol Chem 1999; 274 (47): 33179–33182. doi: 10.1074/jbc.274.47.33179.
11. Kwiatkowski DJ. Functions of gelsolin: motility, signaling, apoptosis, cancer. Curr Opin Cell Biol 1999; 11 (1): 103–108. doi: 10.1016/s0955-0674 (99) 80012-x.
12. de la Chapelle A, Tolvanen R, Boysen G et al. Gelsolin-derived familial amyloidosis caused by asparagine or tyrosine substitution for aspartic acid at residue 187. Nat Genet 1992; 2 (2): 157–160. doi: 10.1016/s0955-0674 (99) 80012-x.
13. Kivela T, Tarkkanen A, Frangione B et al. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja‘s syndrome. Invest Ophthalmol Vis Sci 1994; 35 (10): 3759–3769.
14. Stewart HS, Parveen R, Ridgway AE et al. Late onset lattice corneal dystrophy with systemic familial amyloidosis, amyloidosis V, in an English family. Br J Ophthalmol 2000; 84 (4): 390–394. doi: 10.1136/bjo.84.4.390.
15. Conceicao I, Sales-Luis ML, De Carvalho M et al. Gelsolin-related familial amyloidosis, Finnish type, in a Portuguese family: clinical and neurophysiological studies. Muscle Nerve 2003; 28 (6): 715–721. doi: 10.1002/mus. 10474.
16. Casal I, Monteiro S, Abreu C et al. Meretoja‘s syndrome: lattice corneal dystrophy, gelsolin type. Case Rep Med 2017; 2017 : 2843417. doi: 10.1155/2017/2843417.
17. Huerva V, Velasco A, Sanchez MC et al. Lattice corneal dystrophy type II: clinical, pathologic, and molecular study in a Spanish family. Eur J Ophthalmol 2007; 17 (3): 424–429. doi: 10.1177/112067210701700326.
18. Sunada Y, Shimizu T, Nakase H et al. Inherited amyloid polyneuropathy type IV (gelsolin variant) in a Japanese family. Ann Neurol 1993; 33 (1): 57–62. doi: 10.1002/ana.410330110.
19. Ikeda M, Mizushima K, Fujita Y et al. Familial amyloid polyneuropathy (Finnish type) in a Japanese family: clinical features and immunocytochemical studies. J Neurol Sci 2007; 252 (1): 4–8. doi: 10.1016/j.jns.2006.09.022
20. Ardalan MR, Shoja MM, Kiuru-Enari S. Amyloidosis-related nephrotic syndrome due to a G654A gelsolin mutation: the first report from the Middle East. Nephrol Dial Transplant 2007; 22 (1): 272–275. doi: 10.1093/ndt/gfl548.
21. Gonzalez-Rodriguez J, Ramirez-Miranda A, Hernandez-Da Mota SE et al. TGFBI, CHST6, and GSN gene analysis in Mexican patients with stromal corneal dystrophies. Graefes Arch Clin Exp Ophthalmol 2014; 252 (8): 1267–1272. doi: 10.1007/s00417-014-2648-9.
22. Gorevic PD, Munoz PC, Gorgone G et al. Amyloidosis due to a mutation of the gelsolin gene in an American family with lattice corneal dystrophy type II. N Engl J Med 1991; 325 (25): 1780–1785. doi: 10.1056/NEJM 199112193252505.
23. de la Chapelle A, Kere J, Sack GH Jr. et al. Familial amyloidosis, Finnish type: G654 a mutation of the gelsolin gene in Finnish families and an unrelated American family. Genomics 1992; 13 (3): 898–901. doi: 10.1016/0888-7543 (92) 90182-r.
24. Klaus E, Freyberger E, Kavka G et al. Familial occurrence of a bulbar paralytic form of amyotropic lateral sclerosis with reticular corneal dystrophy and cutis hyperelastica in 3 sisters. Psychiatr Neurol (Basel) 1959; 138 : 79–97.
25. Park KJ, Park JH, Park JH et al. The first Korean family with hereditary gelsolin amyloidosis caused by p.D214Y mutation in the GSN Gene. Ann Lab Med 2016; 36 (3): 259–262. doi: 10.3343/alm.2016.36.3.259.
26. Chastan N, Baert-Desurmont S, Saugier-Veber P et al. Cardiac conduction alterations in a French family with amyloidosis of the Finnish type with the p.Asp187Tyr mutation in the GSN gene. Muscle Nerve 2006; 33 (1): 113–119. doi: 10.1002/mus.20448.
27. Solari HP, Ventura MP, Antecka E et al. Danish type gelsolin-related amyloidosis in a Brazilian family: case reports. Arq Bras Oftalmol 2011; 74 (4): 286–288. doi: 10.1590/s0004-27492011000400012.
28. Cabral-Macias J, Garcia-Montano LA, Perezpena-Diazconti M et al. Clinical, histopathological, and in silico pathogenicity analyses in a pedigree with familial amyloidosis of the Finnish type (Meretoja syndrome) caused by a novel gelsolin mutation. Mol Vis 2020; 26: 345–354.
29. Mattila JS, Krootila K, Kivela T et al. Penetrating keratoplasty for corneal amyloidosis in familial amyloidosis, Finnish type. Ophthalmology 2015; 122 (3): 457–463. doi: 10.1016/j.ophtha.2014.09.035.
30. Bradshaw PJ, Stobie P, Knuiman MW et al. Trends in the incidence and prevalence of cardiac pacemaker insertions in an ageing population. Open Heart 2014; 1 (1): e000177. doi: 10.1016/j.ophtha.2014.09.035.
31. McKenna WJ, Maron BJ, Thiene G. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res 2017; 121 (7): 722–730. doi: 10.1161/CIRCRESAHA.117.309711.
32. Schmidt EK, Atula S, Tanskanen M et al. Causes of death and life span in Finnish gelsolin amyloidosis. Ann Med 2016; 48 (5): 352–358. doi: 10.1080/07853890.2016.1177197.
33. Meretoja J. Genetic aspects of familial amyloidosis with corneal lattice dystrophy and cranial neuropathy. Clin Genet 1973; 4 (3): 173–185. doi: 10.1111/j.1399-0004.1973.tb01140.x.
34. Maury CP, Kere J, Tolvanen R et al. Homozygosity for the Asn187 gelsolin mutation in Finnish-type familial amyloidosis is associated with severe renal disease. Genomics 1992; 13 (3): 902–903. doi: 10.1016/0014-5793 (90) 80072-q.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2021 Issue 5
Most read in this issue
- Analgesic-muscle relaxant infusion in back pain therapy – technological and clinical aspects
- Ofatumumab – a new high-efficacy treatment for relapsing forms of multiple sclerosis
- Ultrasound-guided sacroiliac joint injection
- A syndrome of progressive ataxia and palatal tremor in a patient with mild bilateral idiopathic hypertrophic olivary degeneration