
Epidemiologie, klinický obraz a průběh onemocnění u neuromyelitis optica a onemocnění jejího širšího spektra
Epidemiology, clinical manifestation, and disease course of neuromyelitis optica spectrum disorders
Neuromyelitis optica (NMO) is an autoimmune disease of the CNS and is characterized by typically acute and severe impairment of the optic nerve and spinal cord. Autoantibodies to aquaporin-4 (aquaporin-4 immunoglobulin G; AQP4-IgG) are serological biomarkers of this disease that are detectable in approximately 80% of patients. The assessment of these antibodies has shown that symptoms of this immune-mediated astrocytopathy are not restricted to the optic nerve and spinal cord. Some patients can develop symptoms and signs due to brainstem, diencephalic, and cerebral involvement. The term NMO spectrum disorders (NMOSD) was coined based on these pieces of knowledge. Epidemiological data on this disease are limited but there are some clear differences to multiple sclerosis that may be most commonly considered in the differential diagnosis. The part of AQP4-IgGnegNMOSD patients might have another type of autoantibodies. These antibodies can target the myelin oligodendrocyte glycoprotein. The characteristics of this group of patients are partially different.
Keywords:
optic neuritis – neuromyelitis optica and neuromyelitis optica spectrum disorders – myelitis – area postrema syndrome
Autoři:
J. Libertínová
Působiště autorů:
Neurologická klinika, 2. LF UK a FN Motol, Praha
Vyšlo v časopise:
Cesk Slov Neurol N 2020; 83/116(supplementum 1): 15-19
doi:
https://doi.org/10.14735/amcsnn2020S15
Souhrn
Neuromyelitis optica (NMO) je zánětlivé onemocnění CNS tradičně charakterizované akutním a zpravidla těžkým postižením zrakového nervu a míchy. Sérovým biomarkerem NMO jsou autoprotilátky proti akvaporinu-4 (akvaporin-4 imunoglobulin G; AQP4-IgG), které nacházíme asi u 80 % pacientů. Jejich detekcí se ukázalo, že symptomy této imunitně podmíněné astrocytopatie nejsou omezeny jen na zrakový nerv a míchu. Někteří pacienti s pozitivitou AQP4-IgG mají klinické obtíže vycházející z lézí kmenových, diencefalických i cerebrálních. Vzhledem k celému spektru příznaků byla proto upravena nomenklatura a zaveden jednotící název neuromyelitis optica a onemocnění jejího širšího spektra (neuromyelitis optica spectrum disorders; NMOSD). Epidemiologická data o NMOSD jsou limitována, jsou zde ale některé zjevné odlišnosti proti RS, která je v této souvislosti diferenciálně diagnosticky nejčastěji zvažována. Část AQP4-IgGnegNMOSD pacientů může mít v séru jiný typ protilátek. Tyto jsou často namířené proti myelinovému oligodendrocytárnímu glykoproteinu a charakteristiky této skupiny jsou dílem odlišné.
Klíčová slova:
neuromyelitis optica a onemocnění jejího širšího spektra – optická neuritida – myelitida – syndrom area postrema
Úvod
Neuromyelitis optica (NMO) je zánětlivé onemocnění CNS, které je v tradičním historickém pojetí definované postižením zrakového nervu a míchy, zpravidla s těžkým průběhem a následně významným reziduálním deficitem. Laboratorním biomarkerem je přítomnost protilátek proti vodnímu kanálu, akvaporinu-4 (akvaporin-4 imunoglobulin G; AQP4-IgG), které jsou detekovány asi u 80 % pacientů. S možností jejich detekce se „optikospinální“ fenotyp NMO postupně rozšiřoval a vzhledem k celému spektru možných příznaků byla následně i upravena nomenklatura a zaveden jednotící název NMO a onemocnění jejího širšího spektra (neuromyelitis optica spectrum disorders; NMOSD).
Epidemiologie
Epidemiologické aspekty NMO byly dlouhou dobu opomíjeny a data o incidenci a prevalenci v řadě zemí chyběla. Ve 20. století byla navíc NMO, s ohledem na podobnost klinického obrazu, považována za podtyp RS, a řada pacientů tak byla chybně diagnostikována. Mealy et al ve své analýze uvádějí 29,4 % pacientů s NMOSD původně vedených a léčených pod diagnózou RS [1].
Objevení protilátek AQP4-IgG (dříve také známé jako NMO-IgG) v roce 2004 znamenalo zlom a jasné vymezení NMO od jiných demyelinizačních onemocnění CNS, zejména od RS [2]. Se zavedením rutinního testování autoprotilátek se diagnostika onemocnění výrazně zlepšila a epidemiologických dat začalo přibývat.
Vlivem odlišných metod ke stanovení protilátek, nebo nejednotně užitými diagnostickými kritérii je interpretace těchto dat mnohdy obtížná. Aktuální je podhodnocení incidence a prevalence ve všech studiích publikovaných před poslední revizí a rozšířením diagnostických kritérií z NMO na NMOSD. U některých pacientů s typickým klinickým obrazem NMO navíc AQP4-IgG nenacházíme. Důvody mohou být různé, např. nízká koncentrace protilátek (pod detekčním limitem metody stanovení) v krvi pacienta, nebo odběry provedené až po zahájení léčby.
Tak jako u každého vzácného onemocnění je získání validních epidemiologických parametrů možné jen díky multicentrické a mezinárodní spolupráci a sběru dat v nadnárodních registrech.
Etnická a geografická distribuce
Řada studií shodně poukazuje na vyšší riziko NMOSD ve vztahu k etnickému původu. Předpokládalo se, že onemocnění se častěji vyskytuje v populacích afrických, latinskoamerických či asijských, s relativně vzácným výskytem u populace indoevropské (kavkazské) [3]. V poslední době se nicméně objevují informace, že je prevalence NMOSD u Indoevropanů reálně vyšší, než se dříve předpokládalo [4,5]. Např. Uzawa et al poukazují na přibližně stejnou prevalenci a incidenci NMO po celém světě. Podle něj se spíše liší procentuální podíl NMO v rámci celé skupiny demyelinizačních onemocnění CNS. Je-li prevalence všech demyelinizačních onemocnění CNS (majoritně tvořených RS) u Indoevropanů mnohonásobně vyšší než v Asii [6], pak, ačkoli NMO tvoří jen 1,5 % z nich, je celková prevalence a incidence srovnatelná s těmito parametry v Asii. Tam se NMOSD na počtu demyelinizačních onemocnění CNS podílí z cca 20–40 % [7]. Přesný genetický podklad této skutečnosti zatím není zcela objasněn, ale předpokládá se souvislost s HLA (human leukocyte) antigeny [8,9].
Konkrétně je prevalence NMOSD u indoevropské populace udávána v rozmezí 0,52–4,4 : 100 000 obyvatel (s nejvyšší zaznamenanou hodnotou 4,4 : 100 000 obyvatel v Dánsku) [1]. V ČR je prevalence NMOSD odhadována na 1 : 100 000 obyvatel [7].
Pohlaví a věková distribuce
AQP4-IgGpozNMOSD se dále jednoznačně častěji vyskytuje u žen (8 : 1) [10,11]. Podobně významný nepoměr pohlaví je často popisován i u ostatních protilátkově zprostředkovaných autoimunitních onemocnění [1], která u pacientů s NMO také i častěji nacházíme. Jedná se o systémová onemocnění pojiva, myastenii gravis či idiopatické střevní záněty které mnohdy předchází rozvoji neurologické symptomatiky [11].
K první manifestaci NMOSD může dojít prakticky v kterémkoli věku (jsou popisovány i případy dětí a starších dospělých, sedmou dekádu nevyjímaje [7]), medián věku prvních obtíží ale kolísá mezi 32–41 lety [11,12]. U RS je, pro srovnání, medián věku prvních symptomů cca 24 let a poměr žen a mužů zhruba 2–3 : 1.
Klinický obraz
NMO byla poprvé popsána v 19. století jako monofázické onemocnění postihující zároveň – nebo v těsné návaznosti – oční nerv a míchu. Ve většině (90 %) případů ale toto onemocnění probíhá jako relaps-remitentní (délka remise kolísá od několika týdnů po několik let [7]) a jakkoli jsou ataky očních neuritid a transversálních myelitid jeho základní charakteristikou, mohou se objevovat i jiné symptomy postižení CNS.
Prokázáním AQP4-IgG, jakožto biomarkeru i u jiných než optikospinálních klinických obrazů, se rozšířil fenotyp tohoto onemocnění zejména o symptomy postižení mozkového kmene a hypotalamu [13].
V současné době je pro NMOSD definováno 6 diagnosticky typických klinických obrazů.
Časté jsou:
- optická neuritida;
- akutní myelitida;
- syndrom area postrema.
Méně časté:
- jiný akutní kmenový syndrom;
- symptomatická narkolepsie nebo akutní diencefalický syndrom s NMOSD typickými diencefalickými lézemi na MR;
- symptomatický cerebrální syndrom s lézemi typickými pro NMOSD na MR („ADEM-like“).
Optická neuritida
Optická neuritida (ON) se obecně projevuje bolestí za okem (mnohdy výraznější při pohybu bulbem), snížením zrakové ostrosti, změnou barvocitu či výpadky zorného pole. Symptomatologie ale nemusí být kompletní a existují i případy bez retrobulbární bolesti [7].
ON u NMOSD je obvykle těžká – s rychlým rozvojem i progresí, vedoucí k závažné poruše zraku. Postižení může být oboustranné (někdy současně nebo těsně sekvenčně následující). Zánět typicky postihuje chiasma opticum a v porovnání např. s pacienty s RS zůstává po proběhlém zánětu očního nervu u NMOSD pacientů významný reziduální deficit – amaurózu vyvine po první atace ON cca 22 % pacientů [14].
Klinicky může ON provázet výrazný otok na papile očního nervu. Pomocí vyšetření optickou koherenční tomografií nacházíme po prodělané ON (v odstupu 3 a více měsíců) typicky snížení tloušťky peripapilárních retinálních nervových vláken ve většině kvadrantů zrakového nervu [15].
Myelitida
Myelitida u pacientů s NMOSD probíhá často pod obrazem kompletního míšního syndromu – jako symetrická para/kvadruparéza, resp. plegie s hranicí čití a sfinkterovou poruchou. Na MR (v T2 obrazech) typicky přesahuje 3 obratlové segmenty a bývá proto označována jako longitudinálně extenzivní transverzální myelitida (LETM). Časté je postižení centrální míšní oblasti a preferenčně bývají postiženy segmenty krční a horní hrudní míchy.
Míra úpravy těžkého neurologického deficitu se odvíjí od zahájení adekvátní imunosuprese, nicméně stejně jako u optické neuritidy i zde zůstává často významné reziduum. Riziko trvalého motorického deficitu roste s věkem – u pacientů s počátkem onemocnění po 46. roce bylo více než 40 % následně odkázáno na invalidní vozík (sledování s mediánem 75 měsíců) [16].
Rozsáhlá LETM počínající v krční míše může postupovat směrem k mozkovému kmeni a v důsledku postižení dechového centra v prodloužené míše způsobit neurogenně podmíněnou dechovou tíseň, případně až respirační selhání. Dle některých údajů až třetina pacientů s těžkou transverzální krční myelitidou progredovala bez adekvátní léčby do respiračního selhání s následným úmrtím [17].
Diagnózu NMOSD ale nevylučuje ani klinicky inkompletní míšní léze, jejímž podkladem často bývá myelitida nepřesahující 3 segmenty (tzv. short myelitis), která je považována za příznak typický spíše pro pacienty s RS [18].
S touto myelitidou se můžeme setkat u pacientů léčených imunosupresivní terapií – např. u těch s již diagnostikovanou NMOSD nebo léčených pro jiné autoimunitní onemocnění. Krátká myelitida ale může být i prvním příznakem NMOSD. Flanaganem et al je popisována jako první příznak NMOSD u 14 % pacientů [18]. S ohledem na volbu adekvátní terapie je zde podstatná pečlivá diferenciální diagnostika, kde ve prospěch NMOSD kromě pozitivity AQP4-IgG v séru svědčí i vyšší věk, další autoimunitní onemocnění (a případná imunosuprese) v anamnéze, absence oligoklonálních pásů v likvoru, případně nedostatek dalších nálezů podporujících diagnózu RS.
Až u třetiny pacientů s myelitidou při NMOSD se typicky vyskytují paroxysmální, bolestivé tonické spazmy trvající několik desítek vteřin. Objevují se spíše v období úpravy neurologického deficitu s odstupem několika týdnů od prvních příznaků.
Stejně tak se mohou objevovat radikulární bolesti či Lhermittův příznak.
Naopak jako spíše úvodní příznak rozvíjející se myelitidy u NMOSD bývá popisováno svědění (pruritus) [19,20]. Existují data o pruritu předcházejícímu vznik svalové slabosti při LETM u NMOSD pacientů – u 92 % těchto případů se pruritus nacházel v dermatomech odpovídajících zánětlivému postižení míchy a vzniká pravděpodobně v důsledku zánětlivého postižení zadních rohů míšních [21].
Syndrom area postrema
Z kmenových symptomů se nejčastěji objevuje triáda příznaků – nauzea, zvracení a neztišitelná škytavka – v rámci tzv. syndromu area postrema.
Epizodou těchto obtíží jako prvním příznakem NMOSD dle literatury začíná až 43 % (16–43 %) případů AQP4-IgG séropozitivních pacientů [22]. Stanovení správné diagnózy je tak velkou výzvou. Pacienti bývají přesměrováni ke gastroenterologům a poměrně extenzivně vyšetřováni, podávaná symptomatická terapie je však mnohdy jen s malým efektem. K plné úzdravě naopak dochází po adekvátní imunosupresivní terapii [23].
Jiné kmenové příznaky
Z dalších kmenových syndromů se může při NMOSD objevovat (dle četnosti) diplopie – při okulomotorických dysfunkcích, ztráta sluchu, vertigo či neuralgie trigeminu. Pruritus, interpretován někdy jako symptom postižení zadních provazců míšních, bývá jindy vztahován k postižení periaqueduktálních drah a považován za příznak kmenový.
Riziko postižení dechového centra při rozsáhlých lézích prodloužené míchy, častěji v souvislosti s LETM, již bylo uváděno.
Diencefalické postižení
Hypotalamus je zapojen do regulace cirkadiánního rytmu spánku a bdění, do regulace tělesné teploty, příjmu potravy i řízení autonomního nervového systému.
U pacientů s NMOSD dochází v souvislosti se zánětlivým postižením hypotalamu nejčastěji k dysfunkci jeho hypocretin secernujících neuronů (s následně nízkou koncentrací hypocretinu 1 v mozkomíšním moku). V klinickém obraze se pak objevuje nadměrná denní spavost, hypersomnie (tzv. symptomatická narkolepsie) [24,25]. Klinický obraz je doprovázen přítomností zpravidla bilaterálních T2 hyperintenzit na MR v oblasti hypotalamu.
Zánětlivé postižení hypotalamu se ale může manifestovat i jako jiná autonomní dysfunkce [26,27] – hypotenze, bradykardie, hypotermie, v literatuře byla u pacientů s NMOSD v této souvislosti referována i amenorhea, galactorhea, obezita či diabetes insipidus [7].
Cerebrální syndrom (hemisferální postižení)
Někteří z pacientů s NMOSD vykazují v rámci relapsu onemocnění symptomy encefalopatie – kvalitativní i kvantitativní poruchu vědomí, mohou se vyskytovat i křeče či hemisferální symptomatika (hemiparéza, porucha řeči) [28]. I v tomto případě se někdy může jednat o první manifestaci onemocnění [29]. Na MR je tento stav doprovázen typickými ložisky (atypickými pro RS), svým charakterem podobnými spíše ložiskům při ADEM (ADEM like). U těchto pacientů častěji nacházíme pozitivitu protilátek proti myelinovému oligodendrocytárnímu glykoproteinu (MOG-IgG).
Kožní manifestace
Nespecifickými, méně typickými příznaky pacientů s NMOSD mohou pak být i kožní projevy – bilaterální otoky rukou, erytémy, či z těchto zřejmě nejčastější Raynaudův fenomén [21].
Diagnostická kritéria NMOSD
Ke stanovení diagnózy NMOSD u pacientů s pozitivitou AQP4-IgG stačí přítomnost jednoho ze šesti výše uvedených hlavních klinických projevů.
Neprokážeme-li přítomnost AQP4-IgG, není možné stanovit diagnózu NMOSD na základě jen jednoho klinického symptomu (např. myelitidy) a je třeba alespoň dvou odlišných projevů, přičemž jedním z nich musí být buď optická neuritida, myelitida nebo příznaky z postižení area postrema. Je tak zachován požadavek na diseminaci procesu v prostoru, symptomy mohou být výsledkem jedné nebo více atak.
Žádný z definovaných klinických příznaků není jednoznačně patognomický pouze pro NMOSD. Jejich přítomnost nebo jejich kombinace v kontextu s nálezy na MR by nás však měly vést k indikaci vyšetření AQP4-IgG.
Průběh NMOSD
Příznaky v rámci relapsu NMOSD se většinou vyvíjejí v průběhu několika dnů, postupné zhoršování obtíží po dobu několika měsíců či let je pro NMOSD velmi neobvyklé.
U některých pacientů se optická neuritida a transversální myelitida vyskytují v rámci prvního příznaku současně, monofázicky ale onemocnění probíhá jen cca v 10 % případů (častěji u AQP4-IgG séronegativních pacientů). V ostatních 90 % případů je průběh NMOSD relabující. K relapsu dochází po prvním příznaku u 60 % pacientů do jednoho roku, u 90 % pacientů v průběhu následujících 3 let.
Úprava neurologického deficitu vzniklého při relapsu trvá týdny (měsíce), v rámci NMOSD zůstávají zpravidla významné neurologické deficity, které v důsledku opakovaných relapsů mohou během několika let vést k invaliditě.
Na rozdíl od RS zde nedochází k postupnému zhoršování podobnému sekundárně progresivní fázi RS.
Kromě nálezů typických pro NMOSD je dobré si uvědomit skutečnosti signalizující spíše možnost alternativních diagnóz – „red flags“ (tab. 1).

NMOSD – neuromyelitis optica a onemocnění jejího širšího spektra; OCB – oligoklonální pásy
MOG encefalomyelitis (někdy také anti-MOG syndrom, nebo onemocnění asociovaná s MOG-IgG)
U pacientů s negativitou AQP4-IgG, ale se symptomatologií oční neuritidy, myelitidy, kmenové encefalitidy či akutní diseminované encefalomyelitidy (ADEM-like) je doporučeno stanovení MOG-IgG.
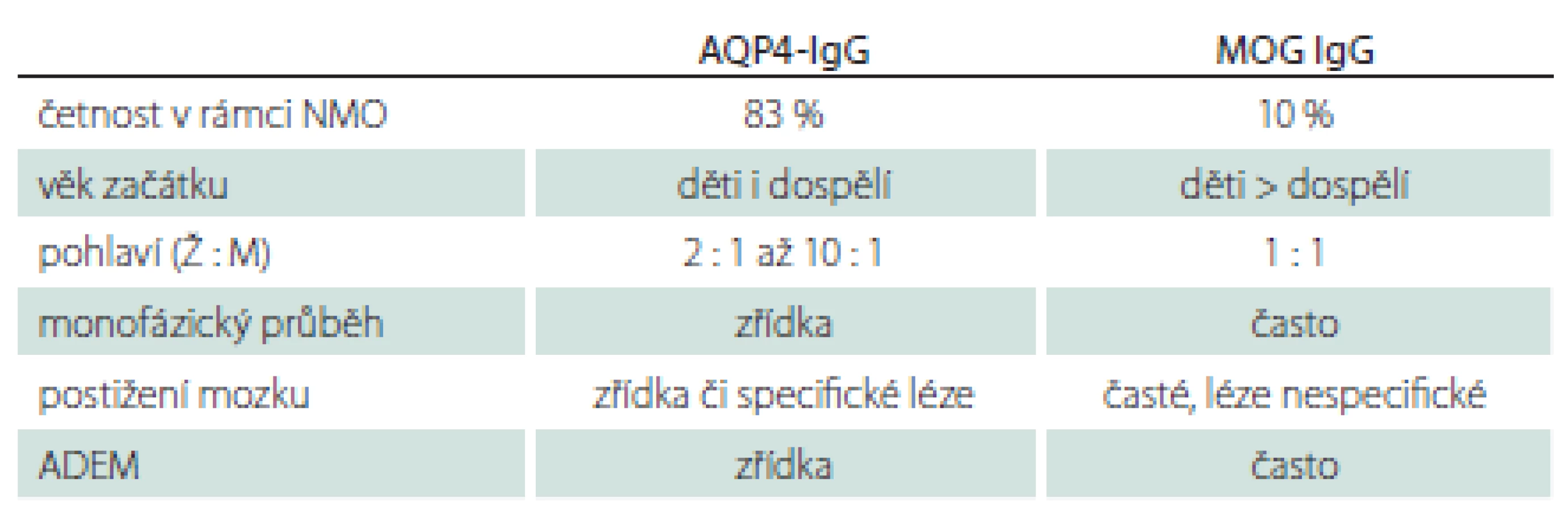
MOG-IgG byly původně etiologicky vztahovány k RS, v poslední době jsou ale MOG encefalomyelitidy považovány za samostatnou jednotku [30]. Fenotypicky i radiologicky se toto onemocnění může překrývat se symptomatologií typickou pro RS i pro NMOSD (řada pacientů může být nesprávně diagnostikována). V porovnání s AQP4-IgGpozNMOSD jsou zde některé odlišnosti (tab. 2) [31]. MOG-IgGpozNMOSD dále charakterizuje dobrá bezprostřední odpověď na terapii intravenozními kortikoidy, stejně jako exacerbace onemocnění po rychlém snížení pokračující perorální kortikoterapie.
Další poruchy asociované s pozitivitou AQP4-IgG
Hypovitaminóza B12
V souvislosti s míšním postižením při NMOSD byly popsány i případy souběžné hypovitaminózy B12 (s pravděpodobně kombinovaným klinickým obrazem).
AQP4 je totiž exprimován i na parietálních buňkách žaludeční sliznice, a protilátky proti AQP4 tak mohou omezit sekreci tzv. intrinsic faktoru s následnou malabsorpcí vitamínu B12 [32,33].
Transientní asymptomatická elevace CK
AQP4 se dále vyskytuje i na sarkolemě svalů, a tak AQP4-IgG může být postižena i svalová membrána. Jsou popisovány případy NMOSD pacientů s myalgiemi a autoimunitní myopatií.
Podobný kontext mají i kazuistiky pacientů s NMOSD a s přechodnou elevací sérové CK („hyperCKémií“) často předcházející, či asociovanou s atakou NMOSD [34].
Závěr
Pacienty s NMOSD dnes definuje podstatně širší spektrum klinických obtíží, než bylo uváděno v klasickém pojetí NMO. „Optikospinální“ paradigma NMO se průkazem AQP4-IgG rozšiřuje zejména o symptomy postižení mozkového kmene (škytavka, zvracení), hypotalamu (hypersomnie) či o syndromy cerebrální. U některých AQP4-IgGneg NMOSD pacientů nalézáme MOG-IgG a s nimi spojené dílčí specifické charakteristiky.
Včasné stanovení správné diagnózy urychluje zahájení adekvátní léčby s následným poklesem kumulativní disability nastupující po opakovaných relapsech. Pacienti s chybně diagnostikovanou RS mohou být léčeni preparáty interferonu beta, které nejenže nepotlačují příznaky NMOSD, ale naopak aktivitu tohoto onemocnění mohou zvyšovat.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
MUDr. Mgr. Jana Libertínová, Ph.D.
Neurologická klinika
2. LF UK a FN Motol
V Úvalu 84
150 06, Praha 5
e-mail: jana.libertinova@fnmotol.cz
Zdroje
1. Mealy MA, Wingerchuk DM, Greenberg BM et al. Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol 2012; 69 (9): 1176–1180. doi: 10.1001/archneurol.2012.314.
2. Lennon VA, Wingerchuk DM, Kryzer TJ et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364 (9451): 2106–2112. doi: 10.1016/S0140-6736 (04) 17551-X.
3. Asgari N. Epidemiological, clinical and immunological aspects of neuromyelitis optica (NMO). Dan Med J 2013; 60 (10): B4730.
4. Asgari N, Lillevang ST, Skejoe HPB et al. A population-based study of neuromyelitis optica in Caucasians. Neurology 2011; 76 (18): 1589–1595. doi: 10.1212/WNL.0b013e3182190f74.
5. Cossburn M, Tackley G, Baker K et al. The prevalence of neuromyelitis optica in South East Wales. Eur J Neurol 2012; 19 (4): 655–659. doi: 10.1111/j.1468-1331.2011.03529.x.
6. Uzawa A, Mori M, Kuwabara S. Neuromyelitis optica: concept, immunology and treatment. J Clin Neurosci 2014; 21 (1): 12–21. doi: 10.1016/j.jocn.2012.12.022.
7. Nytrová P, Horáková D. Neuromyelitis optica. Cesk Slov Neurol N 2015; 78/111 (2): 130–137. doi: 10.14735/amcsnn2015130.
8. Brum DG, Barreira AA, dos Santos AC et al. HLA-DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult Scler 2010; 16 (1): 21–29. doi: 10.1177/1352458509350741.
9. Zéphir A, Fajardy I, Outteryck O et al. Is neuromyelitis optica associated with human leukocyte antigen? Mult Scler 2009; 15 (5): 571–579. doi: 10.1177/13524585 08102085.
10. Marrie RA, Gryba C. The incidence and prevalence of neuromyelitis optica: a systematic review. Int J MS Care 2013; 15 (3): 113–118. doi: 10.7224/1537-2073.2012-048.
11. Nytrova P, Kleinova J, Preiningerova Lizrova J et al. Neuromyelitis optiva a poruchy jejího širšího spektra – retrospektivní analýza klinických a paraklinických nálezů. Cesk Slov Neurol N 2015; 78/111 (1): 72–77. doi: 10.14735/amcsnn201572.
12. Glisson CHC. Neuromyelitis optica spectrum disorders. Available from URL: https: //www.uptodate.com/contents/neuromyelitis-optica-spectrum-disorders.
13. Weinshenkers BG, Wingerchuk DM. Neuromyelitis spectrum disorders. Mayo Clin Proc 2017; 92 (4): 663–679. doi: 10.1016/j.mayocp.2016.12.014.
14. Wingerchuk DM, Hogancamp WF, O‘Brien PC et al. The clinical course of neuromyelitis optica (Devic‘s syndrome). Neurology 1999; 53 (5): 1107–1114. doi: 10.1212/wnl.53.5.1107.
15. Ratchford JN, Quigg ME, Conger A et al. Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology 2009; 73 (4): 302–308. doi: 10.1212/WNL.0b013e3181af78b8.
16. Kitley J, Leite MI, Nakashima I. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 2012; 135 (Pt 6): 1834–1849. doi: 10.1093/brain/aws109.
17. Wingerchuk DM, Lennon VA, Pittock SJ et al. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66 (10): 1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74.
18. Flanagan EP, Weinshenker BG, Krecke KN et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 2015; 72 (1): 81–87. doi: 10.1001/jamaneurol.2014.2137.
19. He Z, Ren M, Wang X et al. Pruritus may be a common symptom related to neuromyelitis optica spectrum disorders. Mult Scler Relat Disord 2017; 13 : 1–3. doi: 10.1016/j.msard.2017.01.011.
20. Netravathi M, Saini J, Mahadevan A et al. Is pruritus an indicator of aquaporin-positive neuromyelitis optica? Mult Scler 2017; 23 (6): 810–817. doi: 10.1177/1352458516665497.
21. Han J, Yang MG, Zhu J et al. Complexity and wide range of neuromyelitis optica spectrum disorders: more than typical manifestations. Neuropsychiatr Dis Treat 2017; 13 : 2653–2660. doi: 10.2147/NDT.S147360.
22. Wingerchuk DM, Banwell B, Bennett JL. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85 (2): 177–189. doi: 10.1212/WNL.0000000000001729.
23. Shosha E, Dubey D, Palace J. Area postrema syndrome: frequency, criteria, and severity in AQP4-IgG-positive NMOSD. Neurology 2018; 91 (17): e1642–e1651. doi: 10.1212/WNL.0000000000006392.
24. Kallollimath P, Gujjar A, Patil M. Symptomatic narcolepsy as a presenting feature of neuromyelitis optica. Ann Indian Acad Neurol 2018; 21 (2): 156–158. doi: 10.4103/aian.AIAN_74_18.
25. Kanbayashi T, Shimohata T, Nakashima I et al. Symptomatic narcolepsy in patients with neuromyelitis optica and multiple sclerosis: new neurochemical and immunological implications. Arch Neurol 2009; 66 (12): 1563–1566. doi: 10.1001/archneurol.2009.264.
26. Crnošija L, Krbot Skorić M et al. Autonomic dysfunction in people with neuromyelitis optica spectrum disorders. Mult Scler 2020; 26 (6): 688–695. doi: 10.1177/1352458519837703.
27. Barun B, Adamec I, Lovrić M et al. Postural orthostatic tachycardia syndrome: additional phenotypic feature of neuromyelitis optica spectrum disorder. Neurol Sci 2014; 35 (10): 1623–1625. doi: 10.1007/s10072-014-1810-9.
28. Kim W, Kim SH, Huh SY et al. Brain abnormalities in neuromyelitis optica spectrum disorder. Mult Scler Int 2012; 2012 : 735486. doi: 10.1155/2012/735486.
29. Kim W, Kim SH, Lee SH et al. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler. 2011; 17 (9): 1107–1112. doi: 10.1177/1352458511404917.
30. Jarius S, Paul F, Aktas O et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation 2018; 15 (1): 134. doi: 10.1186/s12974-018-1144-2.
31. Seze de J. MOG-antibody neuromyelitis optica spectrum disorder: is it a separate disease? Brain 2017; 140 (12): 3072–3075. doi: 10.1093/brain/awx292.
32. Ishii N, Mochizuki H, Takahashi T et al. A case of simultaneous neuromyelitis optica spectrum disorder and subacute combined degeneration. Neurol Sci 2013; 34 (10): 1819–1821. doi: 10.1007/s10072-013-1301-4.
33. Jarius S, Paul F, Ruprecht K et al. Low vitamin B12 levels and gastric parietal cell antibodies in patients with aquaporin-4 antibody-positive neuromyelitis optica spectrum disorders. J Neurol 2012; 259 (12): 2743–2745. doi: 10.1007/s00415-012-6677-1.
34. Sato D, Fujihara K. Atypical presentations of neuromyelitis optica. Arq Neuropsiquiatr 2011; 69 (5): 824–828. doi: 10.1590/s0004-282x2011000600019.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2020 Číslo supplementum 1
Nejčtenější v tomto čísle
- Nálezy na magnetické rezonanci u neuromyelitis optica a onemocnění jejího širšího spektra
- Laboratorní vyšetření u neuromyelitis optica a onemocnění jejího širšího spektra
- Epidemiologie, klinický obraz a průběh onemocnění u neuromyelitis optica a onemocnění jejího širšího spektra
- Diferenciální diagnostika neuromyelitis optica a onemocnění jejího širšího spektra