
Průběh onemocnění asociovaného s tvorbou protilátek proti myelinovému oligodendrocytárnímu glykoproteinu (MOGAD) – soubor pacientů
Autoři:
P. Hanáková 1,2; P. Danhofer 1; M. Dufek 2; J. Šenkyřík 3; M. Komínek 4; P. Dominik 5; H. Ošlejšková 1
Působiště autorů:
Klinika dětské neurologie LF MU a FN Brno
1; I. neurologická klinika LF MU a FN u sv. Anny, Brno
2; Klinika radiologie a nukleární medicíny LF MU a FN Brno
3; Dětská oční klinika LF MU a FN Brno
4; Klinika dětské anesteziologie a resuscitace LF MU a FN Brno
5
Vyšlo v časopise:
Cesk Slov Neurol N 2024; 87(2): 122-131
Kategorie:
Původní práce
doi:
https://doi.org/10.48095/cccsnn2024122
Souhrn
Cílem této práce je podat komplexní přehled týkající se onemocnění asociovaného s protilátkami proti myelinovému oligodendrocytárnímu glykoproteinu (myelin oligodendrocyte glycoprotein antibody-associated disease; MOGAD) a zároveň poskytnout vlastní praktické zkušenosti s diagnostikou a léčbou této nemoci. Prezentujeme soubor sedmi pacientů sledovaných prospektivně na naší klinice od 1/2018 do 2/2023. Průměrný věk dětí v době diagnostiky byl 11,4 ± 2,8 roku. Všichni pacienti splnili v dané době diagnostická kritéria MOGAD. Prezentujeme jednotlivé případy a upozorňujeme na charakter průběhu nemoci a úskalí léčby. Diskutujeme akutní i chronickou terapii a poukazujeme na nutnost dalších prospektivních studií u dětí s MOGAD.
Klíčová slova:
plazmaferéza – optická neuritida – myelitida – myelinový oligodendrocytární glykoprotein – demyelinizační onemocnění CNS
Úvod
V předchozích letech bylo onemocnění asociované s protilátkami proti myelinovému oligodendrocytárnímu glykoproteinu (MOGAD) zahrnuto pod zastřešující pojem „získaná demyelinizační onemocnění“ společně s RS či neuromyelitis optica a onemocnění jejího širšího spektra. MOGAD je nyní považováno za samostatnou klinickou jednotku s individuálním průběhem, imunopatogenezí, odpovědí na terapii a s rozdílnou prognózou. Jeho diagnostika je podmíněna stanovením protilátek proti myelinovému oligodendrocytárnímu glykoproteinu (MOG) MOG-IgG [1–3].
Terminologie užívaná různými autory není jednotná [4–6]. V literatuře se setkáváme s lehce odlišnými názvy (MOG-encefalomyelitidy, MOG-IgG asociovaná onemocnění, MOG-související choroby a jiné). Unijní pediatrické konsorcium MOG vydalo doporučení používat pojem MOGAD (resp. MOG-ab-associated disorders) pro všechny pacienty s prokázanou pozitivitou MOG-IgG. V další části názvu diagnózy by mělo být upřesněno, zda se jedná o monofazické či rekurentní onemocnění a jakým se projevuje fenotypem. Jako příklad takového názvosloví uvádíme „monofazický MOGAD s fenotypem akutní diseminované encefalomyelitidy (ADEM) “ [1]. V rámci následujícího textu užíváme označení MOGAD či MOG-encefalomyelitida.
První zmínky o protilátkách MOG-IgG se v literatuře objevují v 90. letech 20. století, iniciálně zejména ve spojitosti s RS [7,8]. Dříve se ale k testování protilátek používala metoda ELISA, která již dnes vzhledem k riziku falešně pozitivních výsledků doporučována není. Tyto protilátky jsou namířeny proti MOG, jenž je umístěn na extracelulární části cytoplazmatické membrány výběžku oligodendrocytu obalujícího v myelinových vrstvách axon. Ačkoliv tvoří méně než 0,05 % myelinu, je považován za jeden z kruciálních autoantigenů CNS [9].
Tvorba těchto autoprotilátek probíhá periferně (resp. extratekálně). K jejich potvrzení v séru je doporučeno využívat buněčnou assay (cell based assay), která poskytuje méně falešně pozitivních výsledků ve srovnání s metodou ELISA [8]. MOG-IgG pronikají pasivně přes hematoencefalickou bariéru do CNS, kde iniciují inflamatorní změny a vedou k demyelinizaci.
I když mohou někteří pacienti splňovat diagnostická kritéria neuromyelitis optica a onemocnění jejího širšího spektra (neuromyelitis optica spectrum disorder; NMOSD), patogeneze těchto dvou nemocí je odlišná. Zatímco MOGAD je primárně demyelinizační onemocnění s postižením oligodendrocytů, u NMOSD s pozitivními protilátkami proti akvaporinu 4 (NMO AQP4+) se jedná o primární astrocytopatii a demyelinizace je až sekundárním fenoménem [10,11].
Titr autoprotilátek MOG-IgG je vyšší během relapsu než v remisi nemoci. U některých pacientů, zejména s monofazickým průběhem, mohou protilátky MOG-IgG časem zcela vymizet [12]. Přetrvávající pozitivita protilátek představuje faktor přispívající k vyššímu riziku relapsu. Toto zjištění opravňuje vyšetřovat autoinflamatorní imunoglobuliny opakovaně (za 6–12 měsíců) a zvláště pak u pacientů bez chronické imunosuprese [13,14]. Odůvodněna je také kontrola protilátek u pacienta s jejich vstupní negativitou, pokud nadále přetrvává klinické podezření na MOGAD [12]. Zatím není bohužel známo, jak pracovat s hodnotami titru interindividuálně.
Incidence MOGAD je vyšší v dětském věku [15] a u dívek [8], i když Juryńczyk et al. prezentují vyrovnaný poměr pohlaví [16]. Toto onemocnění má tendenci relabovat [17], zpravidla však s lepší výslednou prognózou, než je zaznamenáno u NMO AQP4+ [18]. Odhadované riziko relapsu se pohybuje poměrně v širokém rozmezí 17–57 % [1,17,19,20].
První diagnostická kritéria byla navržena v roce 2018. Zahrnují monofazická či rekurentně probíhající klinická onemocnění, která jsou v korelaci s demyelinizačním nálezem na MR nebo prokázaným elektrofyziologicky. Zároveň je požadována pozitivita protilátek MOG-IgG [6]. Recentně by měla být respektována diagnostická kritéria postulovaná konsorciem odborníků pod vedením Brendy Barnwell [3], jež pracují také s hodnotou výše titru MOG-IgG protilátek. V případě, že je titr nízký (nižší než 1 : 100) a/nebo jsou autoprotilátky přítomny pouze v likvoru, je nutné splnit podpůrná klinická či kritéria MR (tab. 1).
![Diagnostická kritéria MOGAD. Volně přeloženo z [3].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/b26efa56f12a1b6686d122029c0b0256.png)
Fenotypický projev může být pestrý. U dětí bývá nejčastější iniciální prezentací bilaterální optická neuritida (ON) či ADEM. K dalším klinickým projevům patří transverzální myelitida (TM), encefalomyelitida nebo kmenová encefalitida [3,16,18]. Zejména u mladších dětí řadíme mezi nepříznivé fenotypy takzvaný leukodystrophy-like fenotyp [1,21].
Nejčastější první manifestací bývá ON, která je provázená bolestí bulbu, zhoršením zrakové ostrosti až úplnou amaurózou a poruchou barvocitu. Téměř 90 % pacientů má fundoskopicky potvrzený edém optického nervu. Až u poloviny nemocných je v likvoru pozitivní nález mononukleárů (s převahou lymfocytů) a zvýšená celková bílkovina. Pouze v 10–15 % jsou detekovány oligoklonální pásy [16,18]. MR často prokazuje ložiska v oblasti obou optických nervů lokalizovaná nejčastěji v předním segmentu nervu. Bývají delší než u RS a na rozdíl od onemocnění ze spektra NMOSD bývá chiasma opticum ušetřeno. Ložiska jsou hypersignální v T2 váženém obraze, fluid attenuated inversion recovery (FLAIR) a double inversion recovery (DIR) [12]. MR mozku může být zcela v normě nebo může prokazovat četná demyelinizační ložiska, která se však svými znaky odlišují od jiných demyelinizačních chorob, jak poukazují Vaněčková a Nytrová na svém vlastním souboru pacientů. Ložiska bývají rozsáhlejší s nepravidelným tvarem a nepřesnými okraji v T2 váženém obraze i sekvenci FLAIR ve srovnání s RS [22].
Nález na MR mozku mívá typický vývoj v čase, jak popisují a srovnávají Sechi et al. U MOGAD dochází k regresi hypersignálních ložisek v T2 váženém obraze častěji (72 %) než u RS (17 %) a NMOSD (14 %) [23].
Patologický nález vizuálních evokovaných potenciálů (VEP) je přítomen u 70 % nemocných. Stejnou četnost abnormalit lze zaznamenat při vyšetření optickou koherenční tomografií (OCT). Typické jsou významné zúžení retinální vrstvy nervových vláken (retinal nerve fiber layer; RNFL) a úbytek gangliových buněk peripapilárně ve všech segmentech. Tyto nálezy jsou referovány také bez jasné klinické ON. Rozdíly, které nám pomáhají v diferenciální diagnostice ON, jsou uvedeny v tab. 2 [24].
![Diferenciálně diagnostické znaky optické neuritidy [24].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/be411a24522bd1dc62a8f6c422ae5e72.png)
Rozlišujeme terapii akutní ataky a terapii chronickou. Léčba ataky u dětí zahrnuje podání intravenózního methylprednisolonu (IVMP) v dávce 20–30 mg/kg/den, max. 1 g, po dobu 3–5 dní. V podstatě stejná dávka je doporučena u dospělých pacientů. Délka perorálního taperu kortikoidu není přesně definována, záleží na více faktorech vč. věku pacienta, průběhu nemoci a také na zkušenostech lékaře. MOGAD bývá často kortikoresponzivní a u většiny pacientů se doporučuje užití kortikoidů po dobu až 3 měsíců, u některých i déle. Vždy je nutné zvážit nežádoucí účinky kortikoterapie, obzvláště v období růstu a hormonálních změn [18,24]. Jarius et al. referují účinnost IVMP 50 %. Druhou linií je plazmaferéza (PLEX), u které stejní autoři uvádí další podstatné zlepšení u 40 % non-steroidních respondérů [12]. Armangue et al. udávají efekt akutní léčby ataky až 90 %, přičemž ale nehodnotí efekt jednotlivých léčebných modalit [19]. V neposlední řadě je důležité zmínit podání intravenózních imunoglobulinů (IVIG) a kombinace výše uvedených postupů.
Bruijstens et al. [25] doporučují zahájit chronickou terapii při relabujícím průběhu nemoci (obr. 1) [25,26], nicméně je třeba u všech pacientů zvážit prognostické faktory a léčbu vést individuálně, zejména u dětí s těžším deficitem po prvním relapsu, kde je spíše tendence časnější imunosupresi zahájit.
K chronické terapii v první linii patří rituximab (RTX), azathioprin (AZA), mykofenolát mofetil (MMF) nebo měsíčně dávkované IVIG. Kaneko et al. [27] poukazují na zvýšenou hladinu interleukinu 6 (IL-6) u pacientů s MOGAD. Proto není překvapující, že se v další linii také uplatňují inhibitory receptorů IL-6 (tocilizumab, satralizumab), jejichž účinnost a bezpečnost u dětí s MOGAD zatím dokazují spíše jednotlivé kazuistiky [28]. Podstatné je zmínit také právě probíhající studii týkající se účinnosti satralizumabu u pacientů s MOGAD (ClinicalTrials.gov Identifier: NCT05271409). Portfolio léčiv na poli autoimunit CNS se nadále rozšiřuje. V léčbě různých demyelinizačních onemocnění (zejména onemocnění ze spektra NMOSD) se setkáváme stále častěji s monoklonálními protilátkami anti-CD20 (ocrelizumab), anti-CD19 (inebilizumab) či inhibitorem komplementu (eculizumab). Data prokazující účinnost a bezpečnost léků na rozsáhlejších souborech pacientů pod 18 let věku s MOGAD prozatím chybí [29].
Léky užívané u RS (např. interferon beta, glatiramer acetát či natalizumab) nevykazovaly pozitivní efekt [26].
![Zahájení chronické imunosuprese [25,26]. </br>Fig. 1. Initiation of chronic immunosuppression [25,26].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/05e5c29c678e70ba355e5a81c1f4da2c.png)
Soubor pacientů
Prezentován je soubor šesti dívek a jednoho chlapce sledovaných prospektivně na Klinice dětské neurologie LF MU a FN Brno a v RS Centru při I. neurologické klinice LF MU a FN u sv. Anny v Brně od 1/2018 do 2/2023 (tab. 3). Průměrný věk dětí v době diagnostiky byl 11,4 ± 2,8 let (min. 6, max. 16 let). Všichni pacienti měli prokázanou pozitivitu protilátek MOG-IgG. V likvoru byla přítomna oligocytóza u čtyř pacientů. Oligoklonální pásy pouze v likvoru byly prokázány u dvou z nich (likvorologická reakce typ 2), přičemž další dvě dívky měly v likvoru obraz systémového zánětu (resp. likvorologické reakce typ 4). Celkem bylo akutně léčeno 16 atak (z toho 9× byla indikována PLEX a 3× IVIG).
Nejzávažnější komplikací PLEX byla trombóza venae femoralis vlevo po extrakci dialyzačního katetru u pacientky číslo 1 (tab. 3). U dvou pacientek se vyskytl asymptomatický pokles hemoglobinu o více než 20 %. U žádné z nich nebyla hodnota nižší než 95 g/l a vždy došlo při symptomatické terapii k jejímu vzestupu.
Chronická imunosuprese byla zahájena u čtyř dívek a jednoho chlapce, přičemž u dvou z nich již byla úspěšně vysazena (follow-up 12 a 18 měsíců). MR mozku prokázala u tří pacientek postkontrastní sycení optického nervu a pět pacientů mělo také demyelinizační léze v bílé hmotě mozku. Dva pacienti by v průběhu onemocnění sice splnili McDonaldova kritéria RS, nicméně léze na MR nebyly typické, a tudíž diferenciální diagnostika byla ihned od počátku vedena jiným směrem. Detailnější popis souboru je uveden v tab. 3. V další části textu uvádíme vybrané případy.
Pacientka 1
Dívka (tab. 3, pacientka 1) narozená v roce 2011 [30] byla přijata na naši kliniku v 6 letech věku k došetření suspektního získaného demyelinizačního onemocnění. Její rodinná i osobní anamnéza byla bez pozoruhodností.
Od dubna 2018 si stěžovala na horší vizus na levém oku, následně také pravostranně. Fundoskopicky byla potvrzena oboustranná atrofie papil zrakového nervu s temporálním maximem. V likvoru bylo přítomno 10 lymfocytů a normální množství celkové bílkoviny, nenarušená hematoencefalická bariéra a potvrzeny dva identické oligoklonální pásy v likvoru i séru (typ reakce 4). Výsledky sérových autoimunitních faktorů systémových onemocnění (ANA, anti dsDNA, ANCA a jiné) i ostatní laboratorní markery byly v normě. Iniciální MR mozku (obr. 2) prokázala četné asymetrické demyelinizační léze s maximální velikostí jednotlivých ložisek do 11 mm (největší léze v hypotalamu, další dvě paraventrikulárně vpravo, několik juxtakortikálně, zvýšená intenzita optických nervů oboustranně, vše bez postkontrastního sycení). Nález nebyl dle tvaru ložisek zcela typický pro RS. MR míchy byla v normě. Vstupní oční vyšetření zrakové ostrosti potvrdilo dle optotypů vizus pravého oka (OD) 6/60 a levého (OS) 80 cm. Diagnózu bilaterální ON podpořil také patologický nález VEP. V séru byly prokázány pozitivní protilátky MOG-IgG. Dívka byla léčena IVMP (celkem 2,5 g po dobu 5 dní) s následným perorálním taperem kortikoidů (iniciální dávka 1 mg/kg/den prednisonu s pomalou redukcí). Vzhledem k přetrvávajícímu zrakovému deficitu bylo realizováno pět cyklů PLEX a podán 1 g/kg IVIG. Při dimisi přetrvávala mírná alterace vizu na levém oku. Kompletní klinické úpravy a výrazné regrese nálezu v MR (přetrvávalo pouze jedno ložisko paraventrikulárně vpravo) bylo dosaženo během 3 měsíců. Vzhledem k elevaci jaterních testů, výrazné leukocytóze (27×109), anemii a cushingoidnímu habitu byly perorální kortikosteroidy redukovány v rychlejším schématu, než je u MOGAD zvyklostí (tzn. během 6 týdnů namísto 3–6 měsíců). Druhá ataka ON byla relativně záludná. Začala stížnostmi pacientky na bolesti hlavy okcipitálně a subjektivním poklesem zraku na levém oku. Zraková ostrost byla ale objektivně zcela v normě a sektorový lékař odeslal dívku k rehabilitaci s bolestí hlavy z vertebrogenní příčiny. Až po měsíci celkového subjektivního zlepšení byl prokázán levostranný zrakový deficit s centrálním skotomem na levém oku při vyšetření perimetrem. Relaps podpořil nález šesti buněk v likvoru a zvýšený titr MOG-IgG v séru (1 : 320). Strukturální vyšetření mozku i míchy bylo stacionární. OCT prokázala signifikantní bilaterální ztenčení RNFL (vyjma nasálního kvadrantu OD) a úbytek gangliových buněk ve všech kvadrantech bilaterálně. Pacientka byla léčena ve stejném algoritmu jako u první ataky. PLEX byla komplikována žilní trombózou po vytažení dialyzačního katetru z vena femoralis vlevo s nutností tříměsíční antikoagulační terapie.
V rámci chronické léčby bylo podání AZA u pacientky kontraindikováno vzhledem k nízkým hodnotám aktivity enzymu thiopurin methyltransferázy (TPMT) podpořené pozitivní genetickou analýzou heterozygotní varianty genu pro TPMT *3A/*3C. Při kontrolní MR za 5 měsíců na léčbě MMF byla zjištěna izolovaná radiologická aktivita. Po aplikaci IVMP byla převedena na rituximab. Od té doby byla dívka klinicky i radiologicky stabilní a po 2 letech nekomplikovaného průběhu byla zahájena deeskalace terapie (v souladu s aktuálním doporučením – obr. 1).

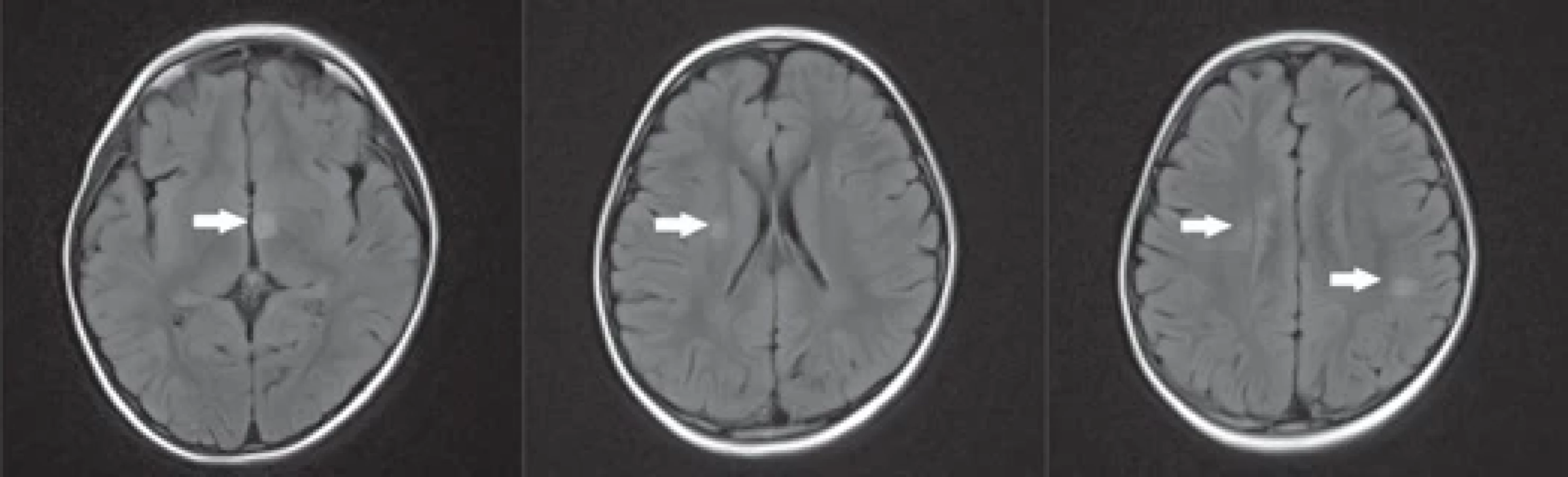
FLAIR – fl uid attenuated inversion recovery

Zároveň byl daný postup výhodný vzhledem k probíhající pandemii COVID-19 a obavám z horšího průběhu této infekce při terapii monoklonální protilátkou anti-CD20 [31].
Aktuálně má ve svých jedenácti letech normální zrakovou ostrost a MR mozku je bez nových demyelinizačních lézí (poslední kontrola proběhla 18 měsíců od vysazení terapie).
Pacientka 2
Dívka (tab. 3, pacientka 2) byla narozená v roce 2007. Matka dívky byla pro blíže neurčené psychiatrické onemocnění zbavena práva péče o dítě, otec byl zdravý. Dívka byla na naši kliniku přeložena v 11 letech věku z jiného pracoviště, kde byla hospitalizována a léčena kortikosteroidy a IVIG pro bilaterální ON asociovanou s tvorbou protilátek MOG-IgG. Do chronické medikace byl zaveden AZA, na kterém se během 4 měsíců rozvinula míšní ataka charakteru parciálního míšního syndromu. Ta již byla léčena nejen IVMP, ale také PLEX. AZA byl následně zaměněn za RTX, při kterém nedošlo ani přes dostatečnou depleci B buněk k remisi onemocnění. Další dvě ataky na této léčbě (ON vpravo a cerebelární syndrom) vyžadovaly kromě léčby 5 g IVMP také provedení série PLEX. V té době se v literatuře objevila studie o prokázaném efektu dlouhodobého podávání IVIG v dávce 1 g/kg/měsíc [26] a pacientka byla na této terapii více než jeden a půl roku kompenzována. Při stabilizaci bylo přistoupeno k pomalé redukci dávky IVIG. V dané době propukla pandemie COVID-19 a otec se rozhodl nechat dívku vakcinovat. Do 2 měsíců se u ní rozvinula bilaterální ON s nutností podání IVMP a bylo rozhodnuto o návratu k iniciální dávce IVIG 1 g/kg/měsíc. Přesto nedošlo ke stabilizaci nemoci a v prosinci 2021 byla opět prokázána ataka pravostranné ON s indikací IVMP vč. perorálního taperu a pěti cyklů PLEX v denním schématu. V rámci chronické medikace byl přidán do dvojkombinace MMF. Při jeho titraci byla nutnost léčit ještě jednu exacerbaci ON. Po roce adekvátního užívání ale pacientka prodělala další ataku ON vpravo, proto byla terapie změněna za subkutánní tocilizumab, na kterém je nyní již rok klinicky stabilizována. Poslední provedená OCT prokázala progresi atrofie disku optických nervů oboustranně (obr. 3).
Obr. 4 souhrnně ilustruje postupný vývoj demyelinizačních změn v MR mozku.



Pacientka 3
Čtrnáctiletá dívka (tab. 3, pacientka 5) byla hospitalizována v prosinci 2021 pro pravostrannou ON. Její otec zemřel při kardiální zástavě v 38 letech věku (s blíže nespecifikovanou kardiomyopatií). Dívka náhlému úmrtí i neúspěšným pokusům o kardiopulmonální resuscitaci zdravotnickými pracovníky přihlížela, což vedlo mimo jiné k její následné trypanofobii. Zavádění intravenózních vstupů a realizace nutných laboratorních odběrů byly pro dívku velmi stresující a tato komplikace ovlivňovala některá naše další rozhodování během hospitalizace. V rodině nikdo netrpěl autoimunitním onemocněním. Dva měsíce před rozvojem ON se u ní nově manifestovaly bolesti hlavy doprovázené zvýšenou únavou. Sama referovala výraznější fyzickou i psychickou zátěž v souvislosti se závodní sezónou ve fotbalu.
Subjektivně byla při přijetí přítomna porucha zraku až charakteru amaurózy na pravém oku. V objektivním nálezu byl přítomen relativní aferentní pupilární defekt vpravo. Při přijetí byla provedena MR, která zobrazila hyperintenzní pravý optický nerv s postkontrastním enhancementem. Ostatní mozková tkáň i celá mícha byly bez demyelinizačních lézí. Oční lékař popsal edém obou papil optických nervů. Likvorologické vyšetření bylo v korelaci s MOGAD. Ihned byla započata terapie IVMP v dávce 30 mg/kg/den s minimálním efektem. Za další dva dny bylo rozhodnuto o provedení čtyř cyklů PLEX v analgosedaci s úplnou úpravou zraku 5. den. Dívka byla převedena na udržovací perorální kortikoterapii po dobu 3 měsíců. Při poslední kontrole (1 rok od ataky) byl její neurologický nález stacionární.
Pacient 4
V prosinci 2022 jsme přijali do Dětské nemocnice 16letého chlapce s těžkou akutní myelitidou (tab. 3, pacient 7). Rozvoji klinických potíží předcházela virová infekce dýchacích cest a při přijetí byl subfebrilní. Jeho starší sestra prodělala v 18 letech CMP, rodinná anamnéza byla bez autoimunit. Vstupní neurologický nález odpovídal syndromu laterálních (velmi těžká paraparéza dolních končetin) a zadních provazců míšních s těžkou sfinkterovou dysfunkcí (vč. nevýbavného análního reflexu a retence moči s nutností zavedení močové katetrizace a následné epicystostomie). Vstupní MR míchy prokázala nález myelitidy v celém míšním rozsahu a na MR mozku byla přítomna mnohočetná demyelinizační ložiska bílé hmoty parietálně, frontálně i temporálně vč. léze v pontu (obr. 5 a 6). V likvoru byl prokázán serózní zánět (46 monocytů, 2 polymorfonukleáry, celková bílkovina 0,59 g/l), oligoklonální pásy nebyly přítomny. Byla zaznamenána pozitivita antiboreliových protilátek ve třídě IgG bez přítomné intratekální syntézy. I přes počáteční nutnost překrytí intravenózními antibiotiky jsme se k diagnóze neuroboreliózy nepřikláněli (CXCL13 negativní).
Vyšetření protilátek MOG-IgG nám umožnilo stanovit diagnózu monofazický MOGAD s fenotypem akutní myelitidy. Vzhledem k velmi těžké pseudochabé paraparéze při přijetí byla časně (tzn. do 5 dní od přijetí; do 9 dní od počátku příznaků) po pulzech IVMP a IVIG zahájena PLEX (pět cyklů denně, poté dvakrát obden). V likvoru jsme potvrdili elevaci IL-6, což nám umožnilo individualizovat terapii a podávat tocilizumab i.v. v dávce 8 mg/kg à 4 týdny. V průběhu hospitalizace musel být pacient opakovaně léčen intravenózními antibiotiky pro nález Pseudomonas aeruginosa v moči. Poslední kontrola pacienta proběhla 4 měsíce od začátku potíží, přičemž objektivně neurologicky přetrvává velmi lehká neomezující centrální paraparéza dolních končetin (s maximálním vyjádřením akrálně). Chlapec je plně soběstačný, bez sfinkterové dysfunkce, aktivně sportuje a chůze je vzdálenostně bez omezení. Kontrolní MR mozku i míchy prokazuje významnou regresi hypersignálních ložisek v T2 váženém obraze a v sekvenci FLAIR.
Diskuze
Na naší klinice aktuálně sledujeme sedm dětí s MOGAD. U pacientky 1 jsme indikovali aplikaci RTX. Jedná se o monoklonální protilátku zaměřenou vůči antigenu CD20, který je charakteristický pro určitá vývojová stadia B lymfocytů. V současné době neexistuje jednotné celosvětově uznávané schéma aplikace RTX u dětí s MOGAD (dle SPC je v ČR stále schválen v rámci autoimunit pro revmatoidní artritidu a polyangiitidu). Terapii jsme řídili dle hodnoty CD19+ lymfocytů, což je v souladu s později publikovaným doporučením Unijního pediatrického konsorcia MOG [25]. Iniciální schéma zahrnovalo RTX v dávce 375 mg/m2 celkem čtyřikrát po týdnu a poté byla přibližně každých 6 měsíců (při znovuobjevení se CD19+ lymfocytů) indikována infuze ve stejné dávce dvakrát po 14 dnech. Tento postup se nám osvědčil k dostatečné depleci B lymfocytů. Během léčby pacientka neprodělala závažnou infekci ani jinou komplikaci.
Jak již bylo zmíněno výše, v praxi se u RTX častěji cytoflowmetricky stanovují buňky CD19+, které lépe odráží aktuální stav B lymfocytů. U pacientky 2 se třetí a čtvrtá ataka vyvinula i přes nulové hodnoty těchto buněk. Podobné zkušenosti uvádí Durozard et al. ve své práci [32].
U pacientky 3 a pacienta 4 jsme v případě akutní terapie ataky zvolili razantnější postup a časně doporučili plazmaferézu jako eskalační léčbu první linie. Jedná se o vysoce účinnou metodu, která má ale své nežádoucí účinky. I přes relativně dobrou prognózu MOGAD s fenotypem ON bylo dokázáno, že axonální ztráta může být těžká [20]. Kompletní zotavení je očekáváno u 56–73 % pacientů [33,34].
Zatímco Magaña et al. podporují brzké provedení PLEX s ohledem na její významný efekt, Savransky et al. neprokázali signifikantní asociaci mezi dobou iniciace PLEX a výsledným outcome [35,36].
Zahájení PLEX není vždy jednoduchým rozhodnutím. Je nutné zvážit přínos i rizika dané metody. Excelentní zlepšení neurologického deficitu pacientky 3 (ve škále Expended Disability Status Scale zlepšení o dva stupně) mezi prvním a pátým cyklem nekomplikované PLEX nás utvrdil ve správnosti indikace eskalace akutní terapie. Nicméně nelze prokázat, zda se nejednalo ještě o doznívající účinek IVMP či přirozený průběh onemocnění, tak jak již diskutují Manguinao et al. v jejich publikaci [37]. K jasnému závěru výše uvedené diskuze by bylo nutné provést randomizovanou kontrolovanou klinickou studii.
U pacienta 4 jsme indikovali jako první druholiniovou léčbu TCZ, s vědomím, že není součástí oficiálních doporučení Unijního pediatrického MOG konsorcia [25]. Přesto vnímáme jeho indikaci za oprávněnou vzhledem k vysoké hladině IL-6 v likvoru. Významný efekt TCZ byl prokázán v metaanalýze srovnání účinnosti biologické léčby u MOGAD, kde procento pacientů bez relapsu při užívání AZA, MMF, RTX, IVIG i TCZ bylo 65 %, 73 %, 66 %, 79 % a 93 %, resp. ve skupině dětí bylo bez relapsu 61 % pacientů na AZA, 69 % na MMF, 53 % na RTX, 84 % na IVIG a skupina pacientů léčených TCZ nebyla dle věku rozdělena [38]. Účinnost i bezpečnost TCZ byla mimo jiné prokázána ve studii Ringelsteina et al. zahrnující také pacienty pod 18 let věku [39].
Závěr
Onemocnění asociované s protilátkami proti myelinovému oligodendrocytárnímu glykoproteinu lze považovat za samostatnou klinickou entitu, která má odlišnou imunopatogenezi i přístup k terapii ve srovnání s RS a onemocněním ze spektra NMOSD. Zejména pacienti s relabujícím průběhem nemoci zasluhují naši zvýšenou pozornost. Postupně jsou formulována diagnostická a terapeutická doporučení, na která musíme dynamicky reagovat. Spektrum cílených léčiv se neustále rozšiřuje, prozatím ale nemáme k dispozici dostatek dat a prospektivních studií u dětských pacientů. V blízké době očekáváme aktualizovaný konsenzus evropských odborníků k chronické léčbě MOGAD.
V našich ordinacích se budeme s touto nemocí setkávat stále častěji. Nejen díky dostupnější diagnostice, ale také širší osvětě zdravotnického personálu.
Etické aspekty
Jedná se o retrospektivní monitoraci souboru pacientů. Studie nepodléhá schválení etickou komisí. Pacienti a jejich rodiče souhlasili s diagnostickým a terapeutickým procesem i publikací výše uvedených dat.
Konflikt zájmů
Autoři deklarují, že v souvislosti s předmětem studie nemají žádný konflikt zájmů.
Zdroje
1. Bruijstens AL, Lechner Ch, Flet-Berliac L et al. E.U. paediatric MOG consortium consensus: part 1 – classification of clinical phenotypes of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol 2020; 29 : 2–13. doi: 10.1016/j.ejpn.2020.10.006.
2. Lana-Peixoto MA, Talim N. Neuromyelitis optica spectrum disorder and anti-MOG syndromes. Biomedicines 2019; 7 (2): 42. doi: 10.3390/biomedicines7020042.
3. Banwell B, Bennett JL, Marignier R et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD panel proposed criteria. Lancet Neurol 2023; 22 (3): 268–282. doi: 10.1016/S1474-4422 (22) 00431-8.
4. Borisow N, Mori M, Kuwabara S et al. Diagnosis and treatment of NMO spectrum disorder and MOG-encephalomyelitis. Front Neurol 2018; 9 : 888. doi: 10.3389/ fneur.2018.00888.
5. Dos Passos GR, Oliveira LM, da Costa BK et al. MOG-IgG--associated optic neuritis, encephalitis, and myelitis: lessons learned from neuromyelitis optica spectrum disorder. Front Neurol 2018; 9 : 217. doi: 10.3389/fneur.2018. 00217.
6. Jarius S, Paul F, Aktas O et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflamm 2018; 15 (1): 134. doi: 10.1186/s12974-018-1144-2.
7. Xiao BG, Linington C, Link H. Antibodies to myelin-oligodendrocyte glycoprotein in cerebrospinal fluid from patients with multiple sclerosis and controls. J Neuroimmunol 1991; 31 (2): 91–96. doi: 10.1016/0165-5728 (91) 90014-x.
8. Lang K, Prüss H. Frequencies of neuronal autoantibodies in healthy controls: estimation of disease specificity. Neurol Neuroimmunol Neuroinflamm 2017; 4 (5): e386. doi: 10.1212/NXI.0000000000000386.
9. Johns TG, Bernard CCA. The structure and function of myelin oligodendrocyte glycoprotein. J Neurochem 1999; 72 (1): 1–9. doi: 10.1046/j.1471-4159.1999.0720 001.x.
10. Ratelade J, Zhang H, Saadoun S et al. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol 2012; 123 (6): 861–872. doi: 10.1007/s00401-012-0986-4.
11. Misu T, Fujihara K, Kakita A et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain J Neurol 2007; 130 (Pt 5): 1224–1234. doi: 10.1093/brain/awm047.
12. Jarius S, Ruprecht K, Kleiter I et al. Mog-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long--term outcome. J Neuroinflamm 2016; 13 (1): 280. doi: 10.1186/s12974-016-0718-0.
13. Jarius S, Ruprecht K, Kleiter I et al. Mog-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: frequency, syndrome specifity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflamm 2016; 13 (1): 279. doi: 10.1186/s12974-016-0717-1.
14. Waters P, Fadda G, Woodhall M et al. Serial anti-myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol 2019; 77 (1): 82–93. doi: 10.1001/jamaneurol.2019.2940.
15. Mol CL, Wong Y, Pelt ED et al. The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults. Mult Scler 2020; 26 (7): 806–814. doi: 10.1177/1352458519845112.
16. Juryńczyk M, Jacob A, Fujihara K et al. Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations. Pract Neurol 2019; 19 (3): 187–195. doi: 10.1136/practneurol-2017-001 787.
17. Pandit L, Mustafa S, Nakashima I et al. MOG-IgG-associated disease has a stereotypical clinical course, asymptomatic visual impairment and good treatment response. Mult Scler J Exp Transl Clin 2018; 4 (3): 2055217318787829. doi: 10.1177/2055217318787829.
18. Ramanathan S, Mohammad S, Tantsis E et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry 2018; 89 (2): 127–137. doi: 10.1136/jnnp-2017-316880.
19. Armangue T, Olivé-Cirera G, Martínez-Hernandez E et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol 2020; 19 (3): 234–246. doi: 10.1016/S1474-4422 (19) 30488-0.
20. Bruijstens AL, Breu M, Wendel EM et al. E.U. paediatric MOG consortium consensus: Part 4 – outcome of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol 2020; 29 : 32–40. doi: 10.1016/j.ejpn.2020.10.007.
21. Hacohen Y, Rossor T, Mankad K et al. „Leukodystrophy-like“ phenotype in children with myelin oligodendrocyte glycoprotein antibody-associated disease. Dev Med Child Neurol 2018; 60 (4): 417–423. doi: 10.1111/dmcn.13649.
22. Vaněčková M, Nytrová P. Patologické nálezy na zobrazení pomocí magnetické rezonance u onemocnění asociovaného s pozitivními protilátkami proti myelinovému oligodendrocytárnímu glykoproteinu. Cesk Slov Neurol N 2022; 85/118 (4): 330–335. doi: 10.48095/cccsnn2022330.
23. Sechi E, Krecke KN, Messina SA et al. Comparison of MRI lesion evolution in different central nervous system demyelinating disorders. Neurology 2021; 97 (11): e1097–e1109. doi: 10.1212/WNL.0000000000012 467.
24. Chen JJ, Pittock SJ, Flanagan EP et al. Optic neuritis in the era of biomarkers. Surv Ophthalmol 2020; 65 (1): 12–17. doi: 10.1016/j.survophthal.2019.08.00.
25. Bruijstens AL, Wendel EM, Lechner CH et al. E.U. paediatric MOG consortium consensus: Part 5 – treatment of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol 2020; 29 : 41–53. doi: 10.1016/j.ejpn.2020.10.005.
26. Hacohen Y, Wong YY, Lechner C et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol 2018; 75 (4): 478–487. doi: 10.1001/jamaneurol.2017.4601.
27. Kaneko K, Sato DK, Nakashima I et al. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: a cross--sectional study and potential therapeutic implications. J Neurol Neurosurg Psychiatry 2018; 89 (9): 927–936. doi: 10.1136/jnnp-2018-317969.
28. Kroenke E, Ankar A, Malani S et al. Refractory MOG--associated demyelinating disease in a pediatric patient. Child Neurology Open 2022; 9 : 2329048X221079093. doi: 10.1177/2329048X221079093.
29. Shi M, Chu F, Jin T et al. Progress in treatment of neuromyelitis optica spectrum disorders (NMOSD): novel insights into therapeutic possibilities in NMOSD. CNS Neurosci Ther 2022; 28 (7): 981–991. doi: 10.1111/cns.13 836.
30. Hanáková P, Ošlejšková H, Šenkyřík J et al. Kazuistika pacientky s optickou neuritidou asociovanou s anti-MOG protilátkami. Vox Pediatrie 2020; 20 (1): 33–34.
31. Boekel L, Wolbink GJ. Rituximab during the COVID--19 pandemic: time to discuss treatment options with patients. Lancet Rheumatol 2021; 4 (3): e154–e155. doi: 10.1016/S2665-9913 (21) 00418-5.
32. Durozard P, Rico A, Boutiere C et al. Comparison of the response to rituximab between myelin oligodendrocyte glycoprotein and aquaporin-4 antibody disease. Ann Neurol 2020; 87 (2): 256–266. doi: 10.1002/ana.25648.
33. Wendel EM, Baumann M, Barisic N et al. High association of MOG-IgG antibodies in children with bilateral optic neuritis. Eur J Paediatr Neurol 2020; 27 : 86–93. doi: 10.1016/j.ejpn.2020.04.002.
34. Eyre M, Hameed A, Wright S et al. Retinal nerve fibre layer thinning is associated with worse visual outcome after optic neuritis in children with a relapsing demyelinating syndrome. Dev Med Child Neurol 2018; 60 (12): 1244–1250. doi: 10.1111/dmcn.13757.
35. Magaña SM, Keegan BM, Weinshenker BG et al. Beneficial plasma exchange response in central nervous system inflammatory demyelination. Arch Neurol 2011; 68 (7): 870–878. doi: 10.1001/archneurol.2011.34.
36. Savransky A, Rubstein A, Rios MH et al. Prognostic indicators of improvement with therapeutic plasma Exchange in pediatric demyelination, Neurology 2019; 93 (22): e2065–e2073. doi: 10.1212/WNL.0000000000008551.
37. Manguinao M, Krysko KM, Maddike S et al. A retrospective cohort study of plasma exchange in central nervous system demyelinating events in children. Mult Scler Relat Disord 2019; 35 : 50–54. doi: 10.1016/ j.msard.2019.07.004.
38. Chang X, Zhang J, Shangru L et al. Meta-analysis of the effectiveness of relapse prevention therapy for myelin-oligodendrocyte glycoprotein antibody-associated disease. Mult Scler Relat Disord 2023; 72 : 104571. doi: 10.1016/j.msard.2023.104571.
39. Ringelstein M, Lindenblatt G, Fischer K et al. Interleukin-6 receptor blockade in treatment-refractory MOG-IgG-associated disease and neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm 2022; 9 (1): e1100. doi: 10.1212/NXI.0000000000001100.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2024 Číslo 2
Nejčtenější v tomto čísle
- Průběh onemocnění asociovaného s tvorbou protilátek proti myelinovému oligodendrocytárnímu glykoproteinu (MOGAD) – soubor pacientů
- Faktory ovlivňující outcome u pacientů se syndromem kaudy equiny
- Neurochirurgické intervence v těhotenství
- Efekt bimanuální senzorické rukavice a unimanuální roboticky asistované terapie na funkci horní končetiny po cévní mozkové příhodě