
Familiární amyloidová polyneuropatie – kazuistika
Familiar Amyloid Polyneuropathy – a Case Report
We present a case of a patient with familial amyloid polyneuropathy. A young patient with gradually progressing symptoms of peripheral neuropathy was initially diagnosed and treated for chronic inflammatory demyelinating polyneuropathy. Subsequent complications resulted in endoscopic examination of the digestive tract. Biopsy showed evidence of amyloid deposits. Subsequent comprehensive examination confirmed hereditary form of amyloid polyneuropathy on the basis of a rare mutation in the transthyretin gene in Val50Ala position. Family history revealed that the mother of the patient was treated for amyloidosis with neuropathic symptoms. Treatment with tafamidis meglumine prevented further disease progression, and was followed by orthotopic liver transplantation. At present, clinical condition of the patient gradually improves with regression of motor and sensory neuropathic symptoms.
Key words:
familial amyloid polyneuropathy – transthyretin – tafamidis meglumine
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
T. Pika 1; P. Látalová 2; H. Hůlková 3,4; H. Vlášková 3; P. Otruba 5; V. Mejzlík 6; V. Ščudla 1,7
Působiště autorů:
Hemato-onkologická klinika LF UP a FN Olomouc
1; Ústav klinické a molekulární patologie, LF UP v Olomouci
2; Ústav dědičných metabolických poruch, 1. LF UK a VFN v Praze
3; Ústav patologie, 1. LF UK a VFN v Praze
4; Neurologická klinika LF UP a FN Olomouc
5; Centrum kardiovaskulární a transplantační chirurgie, Brno
6; III. interní klinika – nefrologická, revmatologická a endokrinologická LF UP a FN Olomouc
7
Vyšlo v časopise:
Cesk Slov Neurol N 2015; 78/111(6): 710-714
Kategorie:
Kazuistika
doi:
https://doi.org/10.14735/amcsnn2015710
Předkládáme popis případu nemocného s familiární formou amyloidové polyneuropatie. Mladý nemocný s postupně progredujícími projevy léze periferních nervů byl zprvu diagnostikován a léčen pro chronickou zánětlivou demyelinizační polyneuropatii. Pro následné komplikace bylo provedeno endoskopické vyšetření zažívacího traktu s bioptickým průkazem amyloidových depozit. Následné komplexní vyšetření potvrdilo hereditární formu transtyretinové amyloidové polyneuropatie na podkladě raritní mutace v genu pro transtyretin na pozici Val50Ala.
Souhrn
Předkládáme popis případu nemocného s familiární formou amyloidové polyneuropatie. Mladý nemocný s postupně progredujícími projevy léze periferních nervů byl zprvu diagnostikován a léčen pro chronickou zánětlivou demyelinizační polyneuropatii. Pro následné komplikace bylo provedeno endoskopické vyšetření zažívacího traktu s bioptickým průkazem amyloidových depozit. Následné komplexní vyšetření potvrdilo hereditární formu transtyretinové amyloidové polyneuropatie na podkladě raritní mutace v genu pro transtyretin na pozici Val50Ala. Anamnesticky, matka nemocného byla léčena pro amyloidózu s neuropatickými projevy. Léčba pacienta preparátem tafamidis meglumine vedla k zabránění další progrese onemocnění a následně byla provedena ortotopní transplantace jater. V současnosti dochází ke klinickému zlepšování stavu nemocného s postupnou regresí motorických a senzitivních neuropatických projevů.
Klíčová slova:
familiární amyloidová polyneuropatie – transtyretin – tafamidis meglumine
Úvod
Amyloidózy tvoří poměrně heterogenní skupinu onemocnění, jejichž společným rysem je ukládání amyloidu – insolubilního bílkovinného materiálu fibrilárního charakteru, který je extracelulárně deponován v tkáních. Doposud bylo identifikováno více než 30 různých amyloidogenních proteinů. V zásadě rozlišujeme amyloidózu dle rozsahu postižení na systémovou a lokalizovanou, dle charakteru přenosu pak na získané a hereditární formy [1]. V našich podmínkách se nejčastěji setkáváme s AL (light‑chain amyloidosis) typem amyloidózy, která náleží do skupiny monoklonálních gamapatií a jejíž příčinou je ukládání monoklonálních lehkých řetězců imunoglobulinu [2]. Hereditární amyloidózy představují přibližně 4 % celkového počtu případů a jsou často vázány na endemické oblasti. Nejčastějším dědičným typem je amyloidóza z depozice mutovaného transtyretinu (TTR, prealbumin). Doposud bylo identifikováno více než 130 mutací v transtyretinovém genu a dle typu příčinné mutace je charakterizován i klinický obraz onemocnění, přičemž nejčastější variantu představuje neuropatické postižení [3]. Náplní předloženého sdělení je prezentace raritního případu nemocného s dědičnou formou TTR amyloidózy.
Popis případu
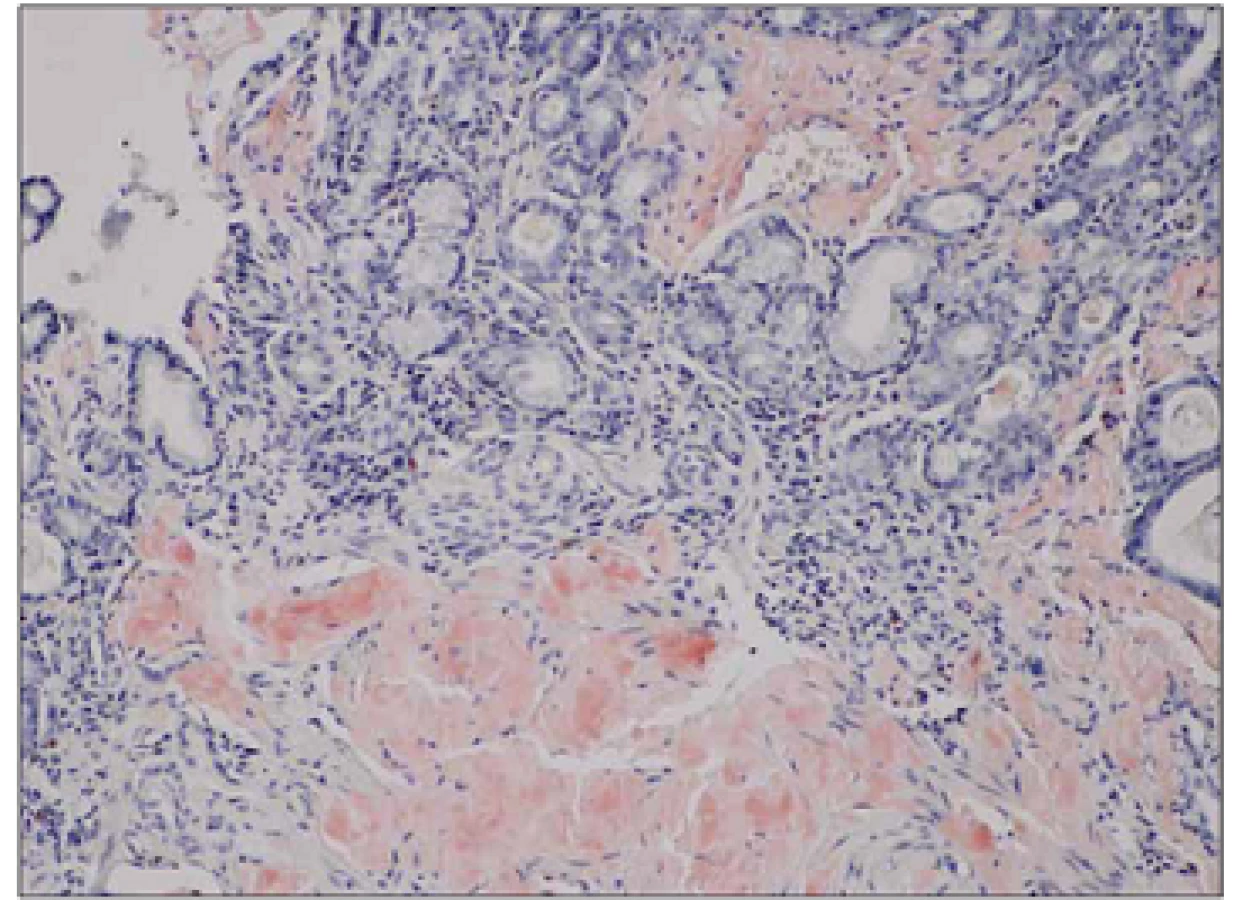
Nemocný (25 let) byl v dubnu 2012 vyšetřen na spádovém neurologickém oddělení pro anamnézu dvou let trvajících a postupně progredujících dysestezií a parestezií dolních končetin spojených s atrofií svalů a omezením svalové síly akrálně pod obrazem peroneální parézy. V posledním roce byla rovněž přítomna intermitentní dyspepsie s úbytkem hmotnosti 10 kg (BMI 16,3; modifikované BMI (mBMI) 717,2). V osobní anamnéze byla zjištěna pouze endoskopicky neverifikovaná refluxní choroba jícnu léčená inhibitorem protonové pumpy. Matka nemocného zemřela ve 47 letech na amyloidózu. Bylo provedeno EMG vyšetření s nálezem těžké axonálně‑demyelinizační senzomotorické polyneuropatie s prevalencí na dolních končetinách. Dlouholatentní odpovědi na nervech dolních končetin (H ‑ reflex a F vlna) nebyly vybaveny, kondukční motorické studie prokazovaly snížení rychlosti motorického vedení na hodnoty mezi 35,3 – 42,9 m/ s. Amplitudy sumačních akčních potenciálů byly snížené s nápadnou chronodisperzí a při vyšetření n. ulnaris až do obrazu bloku vedení. Senzitivní neurogram na dolních končetinách nebyl vybaven, na nervech horních končetin prokazoval sníženou senzitivní rychlost vedení a redukci amplitudy senzitivního nervového akčního potenciálu. Doplněná jehlová EMG prokázala vyšší amplitudu motorických jednotek a výraznou redukci interferenčního vzorce. Tento nález byl interpretován jako změny při chronické zánětlivé demyelinizační polyneuropatii (CIDP) nebo probíhající polyradikuloneuritidě. Vyšetření likvoru ukázalo vyšší hladinu bílkoviny (1,37 g/ l, NR: 0,2 – 0,45 g/ l), jinak bylo zcela negativní, stejně tak další běžná vyšetření neprokázala možnou příčinu postižení (CD ‑ transferin, diabetologické testy, sérologie, paraneoplastické protilátky). Bylo rovněž provedeno elektroforetické vyšetření séra a stanovení sérových hladin volných lehkých řetězců imunoglobulinu s negativním nálezem, což bylo hodnoceno jako dostačující k vyloučení možné amyloidózy. Diagnostickým rozborem byla stanovena diagnóza CIDP, podpořená likvorologickým a EMG vyšetřením, nemocný byl zaléčen pulzy metylprednizonu (3 × 1 g i.v.) následně s udržovací terapií azatioprinem a prednizonem. Nemocný byl předán do domácí péče. Po 10 dnech od dimise byl hospitalizován na psychiatrické klinice pro organickou psychózu s bludy při steroidní terapii, zde během dvou dnů došlo k rozvoji horního dyspeptického syndromu a nemocný byl přeložen na gastroenterologické oddělení. Byla provedena horní endoskopie trávicí trubice s nálezem rozsáhlých nekróz sliznice žaludku, duodena a proximálního jejuna, histologické vyšetření bioptického vzorku prokázalo v žaludeční a duodenální sliznici depozici amyloidu. Nemocný byl přeložen k další péči do Centra pro diagnostiku a léčbu monoklonálních gamapatií, FN v Olomouci. Podrobněji byla doplněna rodinná anamnéza s tím, že matka nemocného zemřela na amyloidózu s neuropatickými projevy, která byla biopticky verifikována z n. suralis. Dědeček nemocného z matčiny strany zemřel v šestém deceninu na blíže nespecifikované neurologické onemocnění. Laboratorní vyšetření neprokázalo přítomnost monoklonálního gradientu v séru a moči, sérové hladiny volných lehkých řetězců byly v mezích normy, stejně tak i srdeční biomarkery (Troponin T, NT ‑ proBNP) byly v mezích normy. Nebyla zjištěna zvýšená proteinurie. Vyšetření kostní dřeně neprokázalo přítomnost monoklonální plazmocelulární populace či jinou atypii. Echokardiografické a MR vyšetření myokardu jednoznačně neprokázalo možné postižení při amyloidóze. AL amyloidóza se tedy zdála jako nepravděpodobná, pro což svědčila i negativní nepřímá imunohistochemie lehkých řetězců kappa a lambda v bioptických vzorcích. Imunohistochemická detekce sérového AA proteinu (SAA) byla v amyloidních depozitech rovněž negativní. Bioptické vzorky byly odeslány ke konzultačnímu vyšetření do laboratoře Ústavu dědičných metabolických poruch, VFN v Praze. Zde provedená imunohistochemická detekce primárních amyloidogenních proteinů prokázala masivní pozitivitu transtyretinu (TTR, prealbumin) v amyloidních depozitech v intersticiu lamina propria mucosae a muscularis mucosae žaludeční a duodenální sliznice. Identický výsledek byl následně potvrzen po zajištění potřebných protilátek i v laboratořích FN Olomouc (obr. 1, 2). Vzhledem k charakteru postižení a rodinné anamnéze byla zvažována hereditární forma amyloidózy, což bylo následně potvrzeno sekvenací TTR genu opět v laboratořích Ústavu dědičných metabolických poruch s průkazem mutace c.149T<C v heterozygotním stavu. Předpokládaným efektem této mutace na protein je záměna valinového za alaninový zbytek na 50. pozici (p.Val50Ala), v maturovaném proteinu se jedná o záměnu na 30. pozici (p.Val 30Ala). Byla tedy potvrzena diagnóza familiární polyneuropatie z depozice amyloidu na bázi transtyretinu s postižením periferního nervového systému a gastrointestinálního traktu. Vzhledem k etiologii onemocnění a dominantní syntéze patologického proteinu byl nemocný indikován k ortotopní transplantaci jater (OLT) jako kauzální terapii. Po doplnění potřebných vyšetření (sérologie, echokardiografie, CT břicha) byl zařazen na čekací listinu k OLT. Vzhledem k rychlé progresi neuropatických obtíží i přes konzervativní terapii (vazodilatancia, vitaminy, rehabilitace, nutriční podpora sippingem) a po prostudování dostupné literatury byla po schválení zdravotní pojišťovnou zahájena terapie preparátem Vyndaqel cps. (Tafamidis meglumine, Pfizer) v dávce 20 mg/ den. Terapie vedla k zastavení další progrese neuropatického postižení, zlepšení nutrice s ústupem dyspeptických obtíží, k nárůstu mBMI a celkovému zlepšení kondice nemocného. Léčba byla komplikována atakou hluboké žilní trombózy vena tibialis pravé dolní končetiny léčené nízkomolekulárním heparinem. V listopadu 2013, tedy 19 měsíců od stanovení diagnózy, byla provedena OLT s dobrým pooperačním průběhem, který byl komplikován časnou kortikosenzitivní rejekční epizodou a bronchopneumonií. V současné době nemocný intenzivně rehabilituje, dochází k postupné regresi motorických a senzitivních neuropatických projevů a zlepšení nutričního stavu nemocného (BMI 17,9; mBMI 841).

Diskuze
Transtyretin (prealbumin) je dominantně syntetizován v játrech, v menší míře pak v plexus choroideus v mozku a v retině, a plní funkci transportního proteinu pro řadu proteinů (tyroxin, retinol) [4 – 6]. Gen TTR kódující transtyretin se nachází 18. chromozomu. Mutace v genu vedou k destabilizaci tetramerů proteinu, který následně získává amyloidogenní potenciál [7]. V současnosti bylo identifikováno více než 130 mutací v genu TTR, s autozomálně dominantním typem dědičnosti. Nejčastější mutace je jednonukleotidová substituce na 30. pozici maturovaného proteinu způsobující záměnu valinu za metionin (p.Val30Met). Jednotlivé mutace se liší v penetraci a dále pak v klinických projevech, které jsou ovlivněny přítomností polymorfizmů či enviromentálními vlivy. Rovněž i prognóza nemocných je závislá na typu mutace a postižených orgánech [4,5,8].
Dominující klinický příznak TTR je pozvolna progredující axonálně‑demyelinizační typ senzomotorické periferní neuropatie, která je přítomna u ~ 90 % nemocných. Výše zmíněná nejčastější mutace p.Val30Met je podkladem pro familiární amyloidovou polyneuropatii I. typu (FAP). Výskyt FAP je vázán dominantně na endemické oblasti, nejčastěji Portugalska (jeden případ na 538 obyvatel), Švédska a Japonska [5]. Klinické příznaky se objevují po druhém deceniu života onemocnění, ale samotný věk klinické manifestace je variabilní. U nemocných v Portugalsku dochází ke klinické manifestaci choroby mezi 20. a 30. rokem s rychlou progresí, naopak u švédských nemocných jsou popisovány klinické projevy až mezi 60. a 70. rokem věku, ačkoliv obě populace nesou identickou mutaci. Tento fenotypový rozdíl je vysvětlován genetickými faktory a enviromentálními vlivy [4,8]. Pro onemocnění FAP je typická vzestupná progredující ztráta citlivosti nejprve dolních a posléze i horních končetin, syndrom karpálního tunelu, bolestivé dysestezie a svalová slabost s následnou atrofií. Postižení autonomního nervového systému je spojeno s ortostatickou hypotenzí, synkopami, impotencí či změnou motility gastrointestinálního traktu. Některá sdělení popisují i postižení centrálního nervového systému (CNS) ve formě leptomeningeální infiltrace vedoucí k ischemickým či krvácivým mozkovým příhodám nebo ataxii [3 – 5,9]. Srdeční postižení bývá pozorováno až u 60 % nemocných s rozvinutým onemocněním. Příznaky zahrnují městnavou srdeční slabost s dušností a otoky, časté jsou arytmie. Echokardiografie prokazuje koncentrickou hypertrofii s rozšířením mezikomorového septa, laboratorně bývá zachycena elevace hladin srdečních biomarkerů [10,11]. Onemocnění s mutacemi v genu TTR (p.Val122Ile, p.Gly47Val) vedoucí k dominantnímu srdečnímu postižení jsou označovány jako familiární amyloidové kardiomyopatie (FAC). Endemickou oblastí pro mutaci p.Val122Ile je oblast Střední Ameriky, vyskytuje se u 3,5 % Afroameričanů [4]. Postižení ledvin, tedy zvýšená proteinurie a/ nebo renální nedostatečnost jsou přítomny u ~ 1/ 3 nemocných a je rovněž závislé na typu příčinné mutace. Je popisována výraznější infiltrace amyloidními hmotami v renální dřeni. Afekce zažívacího traktu se vyskytuje přibližně u poloviny nemocných v době diagnózy onemocnění a závažnost příznaků se stupňuje s progresí choroby. Z klinických příznaků dominuje malabsorpce při infiltraci submukózy amyloidem či poruchách peristaltiky se střídáním průjmu a zácpy. Malnutrice je jedním s nepříznivých prognostických faktorů. Oční postižení zahrnuje zákaly sklivce a zvýšení nitroočního tlaku [5,6].
Mutace identifikovaná u našeho nemocného je velmi vzácná varianta mutace na pozici 30 vyskytující se u FAP. V literatuře byl nález této mutace popsán Jonesem et al v rodině německého původu, jejíž členové trpěli neuropatickými obtížemi a u kterých byla následně FAP nesoucí danou mutaci identifikována. Fenotypově se neliší od časté mutace Val30Met, otázkou však zůstává časnost nástupu projevů onemocnění, jež byla u našeho nemocného v poměrně brzkém věku. Vzhledem k ojedinělému nálezu této mutace je zatím světové písemnictví skoupé na detailnější klinický popis [12].
Samotná diagnostika onemocnění zahrnuje fyzikální vyšetření, detailní rodinnou anamnézu a komplexní neurologické vyšetření s EMG. U nemocných bez známé rodinné anamnézy TTR amyloidózy je potřebné provést histologickou verifikaci amyloidových mas s jejich důslednou typizací pomocí imunohistochemických či proteomických metod. Obvykle bývá prováděna biopsie surálního nervu (diferenciální diagnostika od CIDP) či biopsie z oblasti gastrointestinálního traktu. Doporučována je spolupráce s hematologem k vyloučení mnohem častější formy amyloidózy na podkladě depozice lehkých řetězců imunoglobulinu (AL amyloidóza), což je onemocnění patřící do skupiny plazmocelulárních dyskrazií [2,8]. Definitivní diagnóza je potvrzena sekvenací genu TTR s detekcí patogenní mutace (v České republice provádí Ústav dědičných metabolických poruch, VFN v Praze).
Transplantace jater představuje kauzální léčebnou modalitu, jelikož 95 % TTR je produkováno játry. Představuje standardní terapeutický postup u nemocných schopných OLT podstoupit. První transplantace byla provedena v roce 1990 ve Švédsku u nemocného nesoucího mutaci p.Val30Met. Doposud proběhlo více než 2 000 jaterních transplantací v 19 zemích. Pětileté přežití je uváděno u 82 % nemocných s p.Val30Met mutací, u non‑p.Val30Met je to 59 %. Kratší dlouhodobé přežití u non‑p.Val30Met je vysvětlováno ve vyšší míře pokračující depozicí amyloidu v myokardu po OLT [5,13]. OLT má jednoznačný efekt na neuropatické obtíže, u naprosté většiny nemocných dochází k zastavení progrese neuropatie, u řady pacientů spolu s řádnou podpůrnou a rehabilitační léčbou dochází i k postupné regresi obtíží. Uváděn je rovněž velmi příznivý efekt na nutriční stav nemocných. Naopak progrese depozice TTR v CNS a oku není OLT ovlivněna, jelikož syntéza TTR fyziologicky probíhá i v plexus choroideus a retině. OLT je zatížena celou řadou limitací. Mnoho nemocných není schopno vzhledem k věku, pokročilosti choroby a komorbiditám zákrok podstoupit. Otázkou je i dlouhodobá imunosuprese a možná srdeční progrese nemoci u non‑p.Val30Met variant. OLT by měla být provedena optimálně v časném stadiu onemocnění [5,8,13]. V případě familiární amyloidové kardiomyopatie lze uvažovat o duální transplantaci srdce a jater [13,14]. Vzhledem k tomu, že játra nemocného jsou krom produkce mutovaného TTR většinou plně funkční, je možné ve vybraných případech použít explantovaných jater pro tzv. domino transplantaci pro nemocné s terminálním jaterním postižením, avšak s rizikem pozdějšího rozvoje TTR amyloidózy u příjemce [8].
V současné době je pro léčbu časného stadia FAP registrován lék Vyndaqel (Tafamidis meglumine, 20 mg p.o. denně), jenž stabilizuje tetramery transtyretinu s narušením jeho disociace v monomery a další amyloidogenní složky. Výsledky studií potvrdily efekt na oddálení progrese neuropatických obtíží, zachování nutričního stavu a lepší kvalitu života oproti nemocným užívajícím placebo. Možné použití u jiných forem TTR amyloidózy je zatím předmětem klinických studií (FAC, wtTTR senilní, divoký typ amyloidózy) [5,8,14,15]. Léčba prokázala efekt i u našeho nemocného. Další preparát stabilizující tetramery transtyretinu je diflunisal, který je však stále ve fázi klinického testování. Mezi jiné testované léky patří léky narušující amyloidové fibrily (disruptory) [8,14,16]. Jako nejperspektivnější se jeví kombinace kyseliny tauroursodeoxycholové (TUDCA) s doxycyklinem, klinická studie fáze II nyní probíhá. Předběžné výsledky svědčí pro stabilizující efekt na neuropatické a kardiální postižení při dobré toleranci terapie [17]. Mezi další slibné látky náleží malé interferující RNA (siRNA (ALN ‑ TTR01, ALN ‑ TTR02)) či antisense oligonukleotidy (ISIS ‑ TTR), u kterých preklinické a klinické studie fáze I a II v současnosti probíhají [14,18,19]. Nedílnou součástí terapie je symptomatická léčba k ovlivnění symptomů, nutriční a rehabilitační péče. Multioborová medicínská spolupráce je nezbytná.
Závěr
FAP představuje v našich podmínkách neendemické oblasti raritní onemocnění. Vzhledem k podobnému klinickému obrazu může být zaměněna za jiné, častější klinické jednotky. V případě nejisté diagnózy a zejména rodinné anamnézy neuropatických obtíží je nutné myslet i na možnost FAP. Kauzální terapii představuje OLT, ve vybraných případech lze užít preparát tafamidis meglumine. Další léčebné modality jsou ve fázi klinického testování. Diagnostika a léčba onemocnění vyžaduje multidisciplinární přístup, jak dokresluje naše sdělení.
S podporou grantu NT 12451/ 5, NT 14400, MZ ČR – RVO VFN 64165.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 15. 9. 2015
Přijato do tisku: 15. 10. 2015
MUDr. Tomáš Pika, Ph.D.
Hemato-onkologická klinika
LF UP a FN Olomouc
I. P. Pavlova 6
779 00 Olomouc
e-mail: tomas.pika@fnol.cz
Zdroje
1. Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ et al. Nomenclature 2014: amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014; 21(4): 221 – 224. doi: 10.3109/ 13506129.2014.964858.
2. Ščudla V, Adam Z, Hájek R, Krejčí M, Pika T, Maisnar Vet al. Diagnostika a léčba systémové AL amyloidózy: Doporučení vypracovaná Českou myelomovou skupinou (CMG) a myelomovou sekcí České hematologické společnosti ČLS JEP. Transfuze a hematologie dnes 2013; 19 (Suppl): 60.
3. Ryšavá R. Systémové amyloidózy a jejich léčba. Praha: Maxdorf 2013.
4. Zeldenrust SR. ATTR: diagnosis, prognosis and treatment. In: Gertz MA, Rajkumar SV (eds). Amyloidosis: diagnosis and treatment. New York: Humana Press 2010 : 191 – 204.
5. Sekijima Y. Recent progress in the understanding and treatment of transthyretin amyloidosis. J Clin Pharm Ther 2014; 39(3): 225 – 233. doi: 10.1111/ jcpt.12145.
6. Ueda M, Ando Y. Recent advances in transthyretin amyloidosis therapy. Transl Neurodegener 2014; 3 : 19. doi: 10.1186/ 2047 ‑ 9158 ‑ 3 ‑ 19.
7. Kufová Z, Ševčíková S, Hájek R. Detekce hereditárních amyloidóz. Klin Biochem Metab 2014; 22(2): 65 – 69.
8. Nuvolone M, Obici L, Merlini G. Transthyretin‑associated familial amyloid polyneuropathy – current and emerging therapies. Eur Neurol Rev 2012; 7(1): 14 – 21.
9. Laštovičková J. Hereditární amyloidóza s defektem transthyretinu a její neurologické projevy. Neurol Prax 2011; 12(2): 142 – 144.
10. Arbustini E, Merlini G. Early identification of transthyretin‑related hereditary cardiac amyloidosis. JACC Cardiovasc Imaging 2014; 7(5): 511 – 514. doi: 10.1016/ j.jcmg.2014.03.007.
11. Kristen AV, Scherer K, Buss S, Siepen F, Haufe S, Bauer R et al. Noninvasive risk stratification of patients with transthyretin amyloidosis. JACC Cardiovasc Imaging 2014; 7(5): 502 – 510. doi: 10.1016/ j.jcmg.2014.03.002.
12. Jones LA, Skara JC, Cohen AS, Harding JA, Milunsky A, Skinner M. Familial amyloidotic polyneuropathy: a new transthyretin position 30 mutation (alanine for valine) in family of German descent. Clinin Genet 1992; 41(2): 70 – 73.
13. Suhr OB, Herlenius G, Friman S, Ericzon BG. Liver transplantation for hereditary transthyretin amyloidosis. Liver Transpl 2000; 6(3): 263 – 267.
14. Barreiros AP, Otto G, Kahlen B, Teufel B, Galle PR. Familial amyloidosis: great progress for an orphan disease. J Hepatol 2015; 62 : 483 – 485. doi: 10.1016/ j.jhep.2014.09.008.
15. Coelho T, Maia LF, Martins da Silva A, Cruz MW, Plante ‑ Bordeneuve V, Lozeron P et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012; 79(8): 785 – 792. doi: 10.1212/ WNL.0b013e3182661eb1.
16. Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T et al. Diflunisal trial consortium. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013; 310(24): 2658 – 2667. doi: 10.1001/ jama.2013.283815.
17. Obici L, Cortese A, Lozza A, Lucchetti J, Gobbi M, Palladini G et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid 2012; 19 (Suppl 1): 34 – 36. doi: 10.3109/ 13506129.2012.678508.
18. Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 2013; 369(9): 819 – 829. doi: 10.1056/ NEJMoa1208760.
19. Adams D, Théaudin M, Cauquil C, Algalarrondo V, Slama M. FAP neuropathy and emerging treatments. Curr Neurol Neurosci Rep 2014; 14(3): 435. doi: 10.1007/ s11910 ‑ 013 ‑ 0435 ‑ 3.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2015 Číslo 6
Nejčtenější v tomto čísle
- Nádory očnice
- Novinky ze světa NOAK – „Dienerovo pravidlo 1- 3- 6- 12“ a první antidotum s potvrzeným účinkem
-
Péče o pacienty s dysfagií po cévní mozkové příhodě
Standard léčebného plánu - Psychometrické vlastnosti české verze Epworthské škály spavosti