
A different view on the platelet aggregation inhibitor clopidogrel – a well-suitable anti-oedema agent in a preclinical model of brain injury?
Jiný pohled na inhibitor agregace krevních destiček klopidogrel – vhodné antiedematikum v předklinickém modelu poranění mozku?
Cíl: Neuroprotektivní účinky klopidogrelu již byly prokázány na různých experimentálních modelech. Vzhledem k tomu, že klopidogrel je dobře tolerován a schválen k používání v různých klinických podmínkách, může být atraktivním kandidátem pro další klinická zkoumání, zejména v situaci, kdy se antiedematický účinek jeví jako vhodná adjuvantní strategie, například při poranění mozku. Naším záměrem bylo prozkoumat neuroprotektivní roli klopidogrelu při poranění mozku.
Metody: Pro zkoumání účinků klopidogrelu jsme navodili poranění mozku s použitím modelu chladového traumatu u myší a zkoumali jsme základní mechanizmus přežití/ odumírání buněk s použitím kresolové violeti, barvení TUNEL a analýzy western blot.
Výsledky: Klopidogrel v dávce 3 mg/ kg vedl k signifikantnímu snížení otoku mozku. Podobný pokles byl pozorován při dávce klopidogrelu 10 mg/ kg a 30 mg/ kg. Rovněž jsme prokázali, že klopidogrel blokoval prominentní cesty poškození zánětem a vykazoval silný antiaptotický účinek (3 a 30 mg/ kg), který byl spojen se zvýšenou možností přežití neuronových buněk. Klopidogrel (3, 10 a 30 mg/ kg) měnil dávkově dependentním způsobem hladiny JNK, p-38, AKT, ERK a p53.
Závěr: Naše nálezy prokazují, že klopidogrel může být novým kandidátem pro zmírnění posttraumatického poranění mozku a edému. Domníváme se, že může být aplikován hlavně v akutních fázích cerebrální ischemie, která je charakterizována hemoragickou transformací a edémem mozku.
Klíčová slova:
klopidogrel – poranění mozku – neuroprotektivní účinek – otok mozku – objem infarktu – mechanizmy přežití buněk
Authors:
M. Yalcin Gunal 1,2; B. Yulug 2,3; B. Caglayan 2,4; M. Ozansoy 2,4; U. Kilic 5; I. Keskin 2,6; E. Kilic 2,4
Authors place of work:
Department of Physiology, School of, Medicine, Alanya Alaaddin Keykubat, University, Antalya, Turkey
1; Regenerative and Restorative Medical, Research Center (REMER), University, of Istanbul-Medipol, Istanbul, Turkey
2; Department of Neurology, School of, Medicine, Alanya Alaaddin Keykubat, University, Antalya, Turkey
3; Department of Physiology, School of, Medicine, University of Istanbul-Medipol, Istanbul, Turkey
4; Department of Medical Biology, School of Medicine, University of, Health Sciences, Istanbul, Turkey
5; Department of Histology, School of, Medicine, University of Istanbul-Medipol, Istanbul, Turkey
6
Published in the journal:
Cesk Slov Neurol N 2019; 82(5): 526-532
Category:
Původní práce
Summary
Aim: The neuroprotective effects of clopidogrel have already been shown in various experimental models. Taking into account the fact that clopidogrel is well tolerated and approved for use in various clinical settings, it can be an attractive candidate for further clinical investigations, especially when the anti-oedema effect appears to be a reasonable adjuvant strategy, such as in brain injury (BI). Here we aimed to examine the neuroprotective role of clopidogrel in BI.
Methods: To investigate the effects of clopidogrel, we induced BI in mice using a cold trauma model and evaluated the underlying cell survival/ death mechanisms via cresyl violet, TUNEL staining and western blot analysis.
Results: Clopidogrel at a dose of 3 mg/ kg led to a significant reduction in brain swelling. Similar decreases were observed with 10 mg/ kg and 30 mg/ kg of clopidogrel. We also have shown that clopidogrel blocks the prominent inflammatory injury pathways and exerts a significant anti-apoptotic effect (3 and 30 mg/ kg), which has been associated with increased neuronal cell survival pathways. Clopidogrel (3, 10 and 30 mg/ kg) dose-dependently altered the JNK, p-38, AKT, ERK and p53 levels.
Conclusion: Our findings demonstrate that clopidogrel can be a novel candidate for the reduction of post-traumatic BI and oedema. We propose that it can be applied mainly in the acute phases of cerebral ischaemia, which is characterized by haemorrhagic transformation and brain oedema.
血小板聚集抑制剂氯吡格雷在脑损伤临床前模型中的应用?
目的:氯吡格雷的神经保护作用已经在各种实验模型中得到证实。考考虑到氯吡格雷具有良好的耐受性,并被批准在各种临床环境中使用,它可能是一个有吸引力的临床研究的候选药物,特别是当抗水肿作用似乎是一种合理的辅助策略时,如在脑损伤(BI)中。在此,我们的目的是探讨氯吡格雷在BI中的神经保护作用。
方法:为了研究氯吡格雷对小鼠的影响,我们使用冷损伤模型诱导BI,并通过甲苯酚紫、TUNEL染色和蛋白质印迹分析评估其潜在的细胞存活/死亡机制。
结果:3 mg/ kg剂量的氯吡格雷可显著减轻脑肿胀。氯吡格雷在10 mg/ kg和30 mg/ kg时也有类似的降低。我们还发现氯吡格雷可阻断显著的炎症损伤通路,并发挥显著的抗凋亡作用(3和30 mg/ kg),这与神经元细胞存活通路的增加有关。氯吡格雷(3,10和30 mg/ kg)剂量依赖性地改变了JNK、p-38、AKT、ERK和p53的水平。
结论:我们的研究结果表明,氯吡格雷可以作为减少创伤后双功能障碍和水肿的新候选药物。我们认为它主要应用于以出血性转化和脑水肿为特征的脑缺血急性期。
关键词:氯吡格雷–脑损伤–神经保护作用–脑肿胀–梗死体积–细胞存活途径
Keywords:
clopidogrel – brain injury – neuroprotective effect – brain swelling – infarct volume – cell survival pathways
Introduction
Brain injury (BI) is an important cause of morbidity and mortality and can result in cumulative neurological deficits due to both the initial injury and a series of progressive secondary damages [1,2]. These include various biochemical pathways resulting from the direct physical impairment of neuronal tissue as well as secondary reversible pathobiochemical cascades [3]. The alteration of ionic homeostasis, excitotoxicity, depolarization, mitochondrial impairment and activation of inflammatory and immune cascades that lead to blood-brain barrier (BBB) disruption and related brain oedema have also been reported to play a critical role during the pathogenesis of BI [1–3]. Importantly, recent evidence indicates that high oxidative and inflammatory damage after traumatic BI are associated with early platelet dysfunction and delayed excitotoxic neuronal death [4–9]. This is in line with recent studies showing that oxidative damage and disturbed coagulation play a significant role in the early phases of BI both in humans and animals [10,11]. These findings point out that secondary injury cascades may lead to the development of many of the neurological dysfunctions which are prominent after BI. The delayed nature of BI enables a therapeutic window for neuropharmacological intervention to restrict secondary tissue damage and improve the clinical scores. Therefore, it may allow us to evaluate the secondary injury mechanisms so that new neuroprotective treatment strategies can be developed.
Clopidogrel is a thienopyridine derivative potent antithrombotic drug that inhibits adenosine diphosphate-induced platelet aggregation irreversibly by binding to a P2RY12 purinergic receptor at the platelet surface [12,13]. Recently, it was reported that pre-injury, clopidogrel does not alter the clinical outcomes in BI patients [14], although warfarin has been found to increase mortality in the elderly population with traumatic BI [15,16]. These findings suggest that plasmatic coagulation is a more critical element than platelet function in the prevention of haematoma expansion in acute intracerebral haemorrhage (ICH), which can be a consequence of BI. This is also in line with a recent clinical study (ARMYDA-9 CAROTID) showing that high-dose clopidogrel was significantly effective in reducing the transient ischaemic attack/ stroke rate without increasing the risk of ICH [13].
Given its neuroprotective effects in experimental models and its clinically relevant therapeutic window, clopidogrel is an attractive candidate for further investigation in BI. To investigate the effects of clopidogrel, we induced BI using a cold trauma model, which is a similar model to cerebral ischaemia in respect of its pathophysiological pathways. Based on previous findings showing that clopidogrel exerted significant neuroprotective effects after ischaemic injury, which is characterized by prominent oxidative injury and oedema, we hypothesized that targeting the same pathophysiological cascades may be promising. To this end, we studied the effects of clopidogrel administration in a BI model in mice and analyzed the neurohistological outcomes and oedema volumes 24 h after the induction of BI.
Materials and Methods
Ethical approval
All applicable international, national and/ or institutional guidelines for the care and use of animals were followed. The study was approved by the local ethical committee (approval number: 38828770-604.01.01-E.3988).
Animals
The study was performed at Meditam Research Laboratories of Istanbul Medipol University Twenty-nine male C57BL/ 6 mice, 8–10 weeks old, weighing 25–30 g, were used for the study. The animals were maintained under a constant 12 : 12-h light-darkness regimen (with the lights on daily at 7.00 a. m.) and with ad libitum access to food and water. The mice were housed separately in cages after the operation.
Drug treatment and experimental groups
The mice were randomly divided into four groups. Group 1 (the control group): 5% ethanol in normal saline (N = 7); group 2: clopidogrel 3 mg/ kg (N = 7); group 3: clopidogrel 10 mg/ kg (N = 8) and group 4: clopidogrel 30 mg/ kg (N = 7) [17–19]. All the injections were done intraperitoneally immediately after the experimental procedure.
Cold injury
The brain injury was performed as previously described for a cryogenic trauma model [20,21]. All the mice were anesthetized with intraperitoneal (i.p) ketamine (60 mg/ kg) and xylazine (6 mg/ kg) and fixed in the stereotaxic device. A parietal craniotomy (3 mm diameter, 2.5 mm lateral, 2.5 mm posterior to the bregma) was done using a dental drill. The cold injury was performed using a liquid nitrogen-cooled copper probe (tip diameter 2.5 mm), which was placed on the dura for 60 s and then removed. After that, the scalp was sutured. The rectal temperature was continuously monitored and kept between 36.5 and 37°C with a haemothermic blanket during the procedure. The animals were then taken to the feeding room and the experimenters waited for the animals to recover during the following 24 h post-trauma. At the end of these 24 h, the animals were anesthetized again with high doses of i.p xylazine (20 mg/ kg) and ketamine (100 mg/ kg). The mice were sacrificed and their brains were dissected and put on dry ice. Coronal 18 µm-thick brain sections were taken from the frozen brain tissue for histopathologic and protein analyses.
Cresyl violet staining
The sections were dried at room temperature for 30 min in order to remove the moisture, followed by fixation in a 4% paraformaldehyde solution for 7 min. After washing with distilled water, the sections were placed in a glass chamber containing phosphate buffered saline (PBS) with subsequent shaking of the samples for 5 min at 140 rpm. Then, cresyl violet dye was applied to the sections for 15 min on a shaker with 80 rpm. After staining, the sections were rinsed three times with distilled water and they were dipped into four chambers containing sequentially increasing concentrations of ethanol (70%, 90%, 95% and 100%) for 20 s in each chamber. Finally, xylene was applied to the sections for 3 min at room temperature and the mounting medium was placed onto each slide [22].
Analysis of brain injury
Coronal brain sections from equidistant brain levels, 0.5 mm apart, were stained with Cresyl violet staining according to a standard protocol [23]. On the sections, the border between the injured and non-injured areas was outlined using an image analysis system (Image J; NIH, Bethesta, MD, USA) and the area of the injury was assessed by subtracting the area of the non-lesioned ipsilateral hemisphere from that on the contralateral side. The volume of the injury was calculated by integration of these lesion areas. Oedema was calculated as the volume difference between the ischaemic and the non-ischaemic hemisphere and expressed as a percentage of the intact hemisphere [24].
Analysis of DNA fragmentation
For evaluation of the neuronal injury, the coronal brain sections of the mice were fixed with 4% paraformaldehyde / 0.1 M PBS and were labelled using a TUNEL kit (In Situ Cell Death Detection Kit; Roche, Switzerland). The sections were counterstained with 4΄,6-diamidino-2-phenylindole. The stainings were analyzed by quantifying DNA fragmented cells, each measuring 62,500 μm2, under a confocal microscope (LSM 780, Carl Zeiss, Jena, Germany) in a blinded manner [24].
Analysis of cell survival
For western blot analysis, brain tissue samples were harvested from the injured area. Tissue samples of the same groups were pooled and homogenized with a radioimmunoprecipitation assay lysis buffer (89900, Thermo Fisher Scientific, Waltham, MA, USA) containing a protease and phosphatase inhibitor cocktail (5872, Cell Signalling, Danvers, MA, USA). The total protein content was evaluated using a Qubit 3.0 Fluorometer (Q33216, Invitrogen, Life Technologies Corporation [Carlsbad, CA, USA]) according to the manufacturer’s protocol. Equal amounts of protein (20 µg) were size-fractionated using 4–20% Mini-PROTEAN TGX (4561096, Bio-Rad, Life Sciences Research) gel electrophoresis and then transferred to a PVDF membrane using the Trans-Blot TurboTransfer System (1704155, Bio-Rad, Life Sciences Research, Franklin Township, NJ, USA). Thereafter, the membranes were blocked in 5% non - -fat milk in 50 mMol Tris-buffered saline (TBS) containing 0.1% Tween (TBS-T; blocking solution) for 1 h at room temperature, washed in 50 mMol TBS-T and incubated overnight with polyclonal rabbit phospho-AKT (9275; Cell Signalling), polyclonal rabbit phospho-Erk-1/ -2 (9101; Cell Signalling), polyclonal rabbit phospho-SAPK/ JNK (9251; Cell Signalling), polyclonal rabbit phospho-p38 (9211; Cell Signalling) or polyclonal rabbit phospho-p53 (9284; Cell Signalling). On the next day, the membranes were washed with 50 mM TBS-T and incubated with horseradish peroxidase-conjugated goat-anti-rabbit (sc-2004; Santa Cruz Biotechnology , Dallas, TX, USA) antibody (diluted 1 : 2,500) for 1 h at room temperature. The blots were performed at least three times. Protein loading was controlled by stripping and re-probing with polyclonal rabbit anti-β-actin antibody (4967; Cell Signalling Technology). The blots were developed using a Clarity Western ECL Substrate kit (1705060, Bio-Rad; Life Sciences Research) and visualized using the ChemiDoc MP System (1708280, Bio-Rad; Life Sciences Research). The protein levels were analyzed densitometrically using an image analysis system (Image J; National Institute of Health, Bethesda, MD, USA), corrected with values determined on β-actin blots and expressed as relative values compared with the control group [25].
Statistical analysis
For statistical data comparisons, a standard software package SPSS 18 for Windows (SPSS Inc., Chicago, IL, USA) was used. The differences between groups were analyzed with one-way ANOVA, followed by least significant differences tests. All the values are given as mean ± SD with N values indicating the number of animals analyzed. P Values <0.05 are considered significant.
Results
Infarct Volume and Brain Swelling
To analyze the effects of clopidogrel on BI, the damage volume was measured. In vehicle-treated animals, reproducible brain infarcts were observed after 24 h. In the vehicle-group (group 1), the infarct volume was measured as 26 ± 7.56 mm3 (mean ± SD). For all groups, including the control group, there was no statistically significant difference with regard to the infarct volumes observed (Fig. 1A, B).
Obr. 1. (A) Barvení kresolovou violetí (barvení
Nissl) se běžně používá k identifi kaci
neuronální struktury v mozku. Provádí se za
24 h po poranění mozku. (B) Objem infarktu
a (C) otok mozku. 24 h po poranění mozku
léčba klopidogrelem nezměnila signifikantně rozvoj objemu infarktu. Avšak 3, ale
zejména 10 a 30 mg/kg aplikovaného klopidogrelu
signifi kantně snížilo otok mozku.
Hodnoty jsou uvedeny jako průměr ± SD.
Distanční lišta = 2 mm.
*p < 0,05; **p < 0,01 v porovnání s kontrolní
skupinou, které bylo podáváno vehikulum
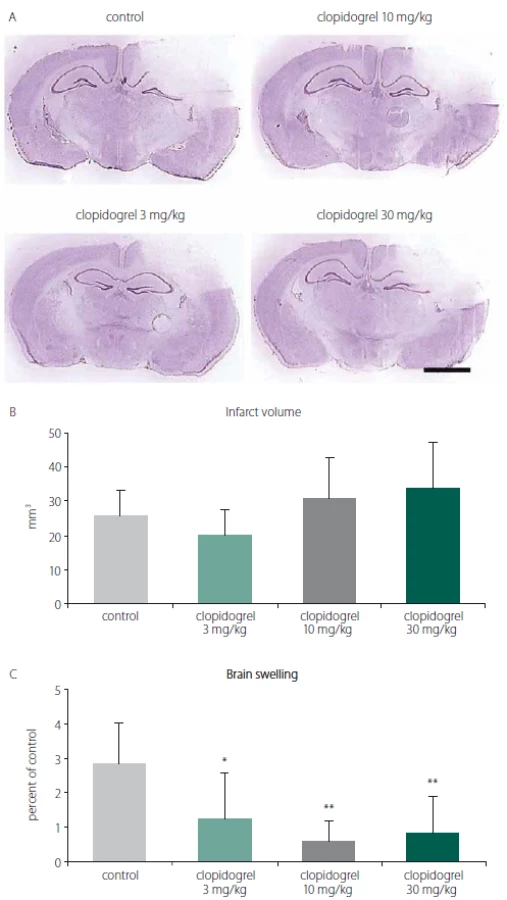
When the alterations of brain swelling were analyzed, however, we observed statistically significant differences. In group 2, where 3 mg/ kg clopidogrel was used, a significant reduction in brain swelling was found (55.98%; P < 0.01). Likewise, significant decreases of brain oedema were also observed in groups 3 and 4, where 10 mg/ kg and 30 mg/ kg clopidogrel, resp., were applied (78.52% and 70.42%; P < 0.05) (mean ± SD) (Fig. 1C).
TUNEL staining and DNA fragmentation
TUNEL staining showed that clopidogrel reduced DNA fragmentation significantly at 3 mg/ kg doses compared to the control group (Fig. 2).
*P < 0.05; **P < 0.01 compared with vehicle-treated control group
Obr. 2. Barvení TUNEL a fragmentace DNA. Barvení TUNEL prokázalo, že klopidogrel
v dávkách 3 mg/kg signifi kantně snižoval fragmentaci DNA v porovnání s kontrolní skupinou.
Hodnoty jsou uvedeny jako průměr ± SD.
*p < 0,05; **p < 0,01 v porovnání s kontrolní skupinou, které bylo podáváno vehikulum

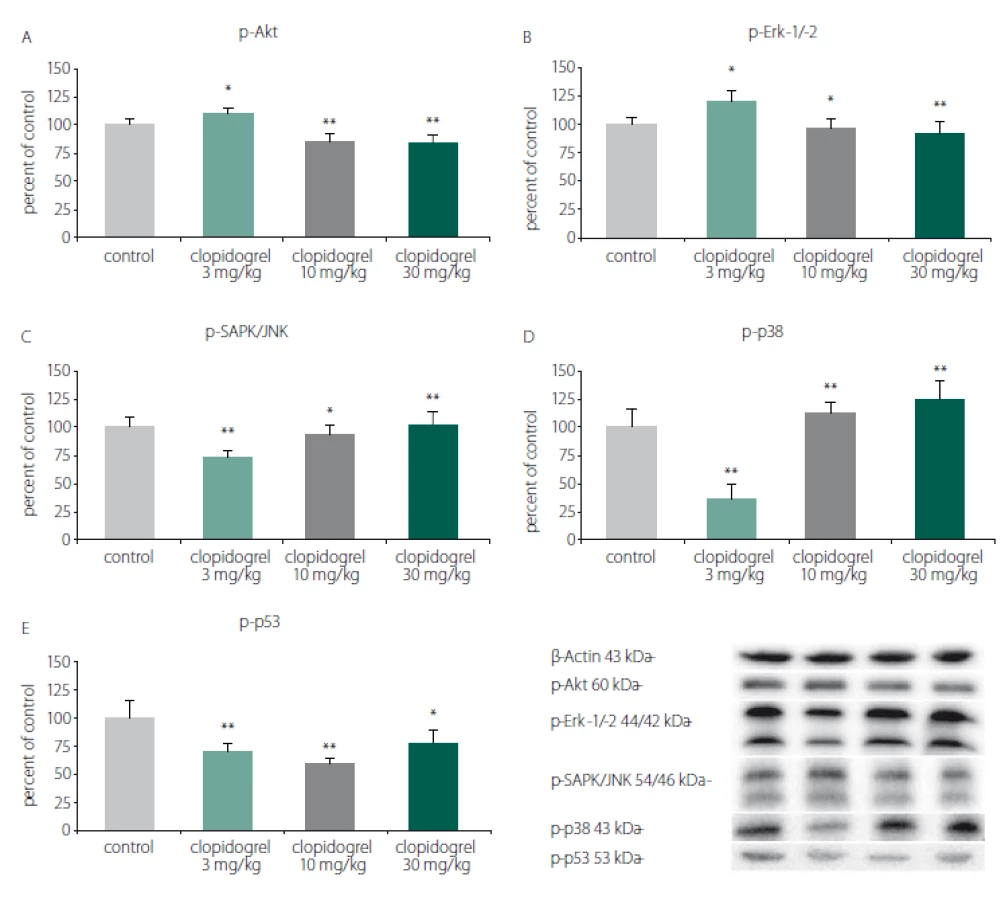
Cell death and survival pathways
Western blot analysis showed that clopidogrel (3, 10 and 30 mg/ kg) dose-dependently altered the c-Jun N-terminal kinases (JNK), p-38 levels, serine/ threonine protein kinase B (AKT), extracellular signal-regulated kinases (ERK), and p53 levels (Fig. 3).
Obr. 3. Mechanizmy odumírání a přežití buněk. (A) p-Akt, (B) p-Erk-1/-2, (C) p-SAPK/JNK, (D) p-p38, (E) p-p53. Analýza western blot prokázala,
že klopidogrel (3, 10 a 30 mg/kg) měnil dávkově dependentním způsobem hladiny JNK, p-38, AKT, ERK a p53. Hodnoty jsou
uvedeny jako průměr ± SD.
*p < 0,05; **p < 0,01 v porovnání s kontrolní skupinou, které bylo podáváno vehikulum
AKT – serin/treonin proteinkináza B; ERK – kináza regulovaná extracelulárním signálem; JNK – c-Jun N-terminální kináza
Discussion
Antiplatelet therapy for the treatment of ischaemic cerebrovascular and cardiovascular diseases is common in the aging population. Due to the accumulating evidence showing the clinical and preclinical anti-inflammatory and anti-oxidative effect of clopidogrel [18,26–36], it became of the utmost importance to know whether clopidogrel therapy exacerbates BI through increasing the haematoma volume after head trauma. It is well-known that BI is a heterogeneous disease process with a complex series of pathophysiological events including contusion necrosis, excitotoxicity, axonal injury, BBB disruption, vasogenic oedema and neuroinflammation [1–11]. From the clinical point of view, previous studies have revealed conflicting results regarding the detrimental effect of the pre-injury use of clopidogrel in BI patients [14,37–39]. For instance, a recent clinical study has revealed that pre-injury brain atrophy, but not (rather than) antithrombotic agent use is correlated with BI-related intracranial haemorrhage [14]. This study indicated that the usage of the antithrombotic agent did not affect the clinical outcome of ICH in the elderly population [38].
Accordingly, preclinical evidence came from experimental studies showing that clopidogrel exerts significant antioxidant and anti-inflammatory activity in various models including ischaemic BI, diabetic ischaemic retinopathy, decompression sickness, experimentally induced peripheral vascular injury and renal ischaemia-reperfusion injury [14,18,26–39]. These studies have indicated that microglial inhibition, inhibition of inflammatory cytokine expression and the infiltration of macrophages as well as the production of matrix metalloproteinases play an essential role in the antioxidant and anti-inflammatory effects of clopidogrel [18,26–36]. Furthermore, clopidogrel has recently been reported to prevent ischaemic reperfusion damage via reducing the nitric oxide levels in the ischaemia-reperfusion model [27]. Accordingly, clopidogrel significantly prolonged the time to produce thrombotic occlusion of the middle cerebral artery and prevented the development of diabetic retinal ischaemia in a streptozotocin-diabetic rat model [18,33]. Highlighting the anti-inflammatory effect of clopidogrel, recent studies have shown that clopidogrel may also ameliorate intimal hyperplasia and attenuate vascular inflammation-related aneurysm formation in various experimental vascular injury models [26,30,34]. The beneficial effects of clopidogrel have been suggested with a recent experimental study indicating that it did not aggravate ICH volume.
It is worth mentioning here that our results indicate a discrepancy between oedema and damage volumes, which seemed to be dose-independent. It is difficult to estimate what caused this difference, but it can be hypothesized that the difference could be related to the heterogeneity of the disease process in BI compared with pure cerebral ischaemia and haemorrhage [40–42]. However, it should also be noted that our decreased vasogenic oedema is an important post-BI predictor of mortality, which also plays a critical pathophysiological role in cerebral ischaemia, haemorrhage and secondary phases of ischaemia.
Since clopidogrel’s anti-oedema effect was not associated with increased haemorrhagic transformation, and our model is mainly adopted as an experimental model of vasogenic brain oedema, our findings could support the acute therapeutic role of clopidogrel not only in BI but also in haemorrhagically transformed ischaemic injury (i.e., haemorrhagic stroke). We have also shown that clopidogrel has exerted a significant anti-apoptotic effect (3 and 30 mg/ kg), which was associated with increased neuronal cell survival pathways. Here, clopidogrel (3, 10 and 30 mg/ kg) dose-dependently altered the JNK, p-38, AKT, ERK and p53 levels in favor of cell survival, which has already been shown to play a critical role in cell death and survival mechanisms [24]. Beyond revealing the dose-independent anti-oedema effect of a well-known antiplatelet agent in an experimental brain trauma model, our findings might suggest that the increased bleeding tendency of clopidogrel may be outweighed by its anti-oedema and neuroprotective effects. This is in accordance with a previous neuroproteomic work stating that acetylsalicylic acid and clopidogrel separately upregulated neuroprotective cellular pathways in experimental traumatic BI [19].
Taken together, our findings could give further insights into the regulation of the secondary prophylaxis treatment for cerebrovascular disease in a specific group of stroke patients who have had an acute BI. In the foreseeable future, further randomized clinical studies evaluating the underlying mechanisms of the clinical neuroprotective effect of clopidogrel in BI are warranted. This might open up a new translational therapeutic window in BI treatment.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Dr. Mehmet Yalcin Gunal
Department of Physiology
School of Medicine
Alanya Alaaddin Keykubat University
Konya Çimento Caddesi No:80
07425 Antalya
Turkey
e-mail: yalcin.gunal@alanya.edu.tr
Accepted for review: 18. 3. 2019
Accepted for print: 16. 7. 2019
Zdroje
1. Park E, Bell JD, Baker AJ. Traumatic brain injury: can the consequences be stopped? CMAJ 2008; 178(9): 1163–1170. doi: 10.1503/ cmaj.080282.
2. Malkesman O, Tucker LB, Ozl J et al. Traumatic brain injury – modeling neuropsychiatric symptoms in rodents. Front Neurol 2013; 4 : 157. doi: 10.3389/ fneur.2013.00157.
3. Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 2010; 31(12): 596–604. doi: 10.1016/ j.tips.2010.09.005.
4. Kochanek PM, Jackson TC, Ferguson NM et al. Emerging therapies in traumatic brain injury. Semin Neurol 2015; 35(1): 83–100. doi: 10.1055/ s-0035-1544237.
5. Lozano D, Gonzales-Portillo GS, Acosta S et al. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat 2015; 11 : 97–106. doi: 10.2147/ NDT.S65815.
6. Angeloni C, Prata C, Dalla Sega FV et al. Traumatic brain injury and NADPH oxidase: a deep relationship. Oxid Med Cell Longev 2015; 2015 : 370312. doi: 10.1155/ 2015/ 370312.
7. Hiebert JB, Shen Q, Thimmesch AR et al. Traumatic brain injury and mitochondrial dysfunction. Am J Med Sci 2015; 350(2): 132–138. doi: 10.1097/ MAJ.0000000000000506.
8. Tataranno ML, Perrone S, Buonocore G. Plasma biomarkers of oxidative stress in neonatal brain ınjury. Clin Perinatol 2015; 42(3): 529–539. doi: 10.1016/ j.clp.2015.04.011.
9. Rochfort KD, Cummins PM. The blood-brain barrier endothelium: a target for pro-inflammatory cytokines. Biochem Soc Trans 2015; 43(4): 702–706. doi: 10.1042/ BST20140319.
10. Donahue DL, Beck J, Fritz B et al. Early platelet dysfunction in a rodent model of blunt traumatic brain injury reflects the acute traumatic coagulopathy found in humans. J Neurotrauma 2014; 31(4): 404–410. doi: 10.1089/ neu.2013.3089.
11. Castellino FJ, Chapman MP, Donahue DL et al. Traumatic brain injury causes platelet adenosine diphosphate and arachidonic acid receptor inhibition independent of hemorrhagic shock in humans and rats. J Trauma Acute Care Surg 2014; 76(5): 1169–1176. doi: 10.1097/ TA.0000000000000216.
12. Siller-Matula J, Schror K, Wojta J et al. Thienopyridines in cardiovascular disease: focus on clopidogrel resistance. Thromb Haemost 2007; 97(3): 385–393.
13. Patti G, Tomai F, Melfi R et al. Strategies of clopidogrel load and atorvastatin reload to prevent ischemic cerebral events in patients undergoing protected carotid stenting. Results of the randomized ARMYDA-9 CAROTID (Clopidogrel and Atorvastatin Treatment During Carotid Artery Stenting) study. J Am Coll Cardiol 2013; 61(13): 1379–1387. doi: 10.1016/ j.jacc.2013.01.015.
14. Dunham CM, Hoffman DA, Huang GS et al. Traumatic ıntracranial hemorrhage correlates with preinjury brain atrophy, but not with antithrombotic agent use: a retrospective study. Plos One 2014; 9(10): e0142287. doi: ARTN e10947310.1371/ journal.pone.0109473.
15. Lauer A, Schlunk F, Van Cott EM et al. Antiplatelet pretreatment does not increase hematoma volume in experimental intracerebral hemorrhage. J Cereb Blood Flow Metab 2011; 31(8): 1736–1742. doi: 10.1038/ jcbfm.2011.22.
16. Foerch C, Arai K, Jin G et al. Experimental model of warfarin-associated intracerebral hemorrhage. Stroke 2008; 39(12): 3397–3404. doi: 10.1161/ STROKEAHA.108.517482.
17. Herbert JM, Tissinier A, Defreyn G et al. Inhibitory effect of clopidogrel on platelet adhesion and intimal proliferation after arterial injury in rabbits. Arterioscler Thromb 1993; 13(8): 1171–1179.
18. De La Cruz P, Arrebola M, Gonzalez-Correa A et al. Effects of clopidogrel and ticlopidine on experimental diabetic ischemic retinopathy in rats. Naunyn Schmiedebergs Arch Pharmacol 2003; 367(2): 204–210. doi: 10.1007/ s00210-002-0657-4.
19. Abou-Abbass H, Bahmad H, Abou-El-Hassan H et al. Deciphering glycomics and neuroproteomic alterations in experimental traumatic brain injury: Comparative analysis of aspirin and clopidogrel treatment. Electrophoresis 2016; 37(11): 1562–1576. doi: 10.1002/ elps.201500583.
20. Hermann DM, Kilic E, Kugler S et al. Adenovirus-mediated glial cell line-derived neurotrophic factor (GDNF) expression protects against subsequent cortical cold injury in rats. Neurobiol Dis 2001; 8(6): 964–973. doi: 10.1006/ nbdi.2001.0448.
21. Keskin I, Gunal MY, Ayturk N et al. Dose-dependent neuroprotective effect of enoxaparin on cold-induced traumatic brain injury. Neural Regen Res 2017; 12(5): 761–764. doi: 10.4103/ 1673-5374.206646.
22. Qu Y, Van der Gucht E, Massie A et al. In vivo microdialysis in the visual cortex of awake cat. III: histological verification. Brain Res Brain Res Protoc 2001; 7(1): 52–60.
23. Alvarez-Buylla A, Vicario DS. Simple microcomputer system for mapping tissue sections with the light microscope. J Neurosci Methods 1988; 25(2): 165–173.
24. Kilic U, Caglayan AB, Beker MC et al. Particular phosphorylation of PI3K/ Akt on Thr308 via PDK-1 and PTEN mediates melatonin‘s neuroprotective activity after focal cerebral ischemia in mice. Redox Biol 2017; 12 : 657–665. doi: 10.1016/ j.redox.2017.04.006.
25. Beker MC, Caglayan AB, Kelestemur T et al. Effects of normobaric oxygen and melatonin on reperfusion injury: role of cerebral microcirculation. Oncotarget 2015; 6(31): 30604–30614. doi: 10.18632/ oncotarget.5773.
26. Coimbra LS, Steffens JP, Rossa C Jr et al. Clopidogrel enhances periodontal repair in rats through decreased inflammation. J Clin Periodontol 2014; 41(3): 295–302. doi: 10.1111/ jcpe.12203.
27. Kanko M, Ozden M, Maral H et al. Effect of clopidogrel on nitric oxide levels in an ischemia reperfusion model. J Cardiovasc Pharmacol 2006; 48(1): 797–801. doi: 10.1097/ 01.fjc.0000211795.45281.9d.
28. Kanko M, Maral H, Akbas MH et al. Protective effects of clopidogrel on oxidant damage in a rat model of acute ischemia. Tohoku J Exp Med 2005; 205(2): 133–139.
29. Webster CM, Hokari M, McManus A et al. Microglial P2Y12 deficiency/ inhibition protects against brain ischemia. PLoS One 2013; 8(8): e70927. doi: 10.1371/ journal.pone.0070927.
30. Liu O, Jia L, Liu X et al. Clopidogrel, a platelet P2Y12 receptor inhibitor, reduces vascular inflammation and angiotensin II induced-abdominal aortic aneurysm progression. PLoS One 2012; 7(12): e51707. doi: 10.1371/ journal.pone.0051707.
31. Hu H, Batteux F, Chereau C et al. Clopidogrel protects from cell apoptosis and oxidative damage in a mouse model of renal ischaemia-reperfusion injury. J Pathol 2011; 225(2): 265–275. doi: 10.1002/ path.2916.
32. Bao XC, Chen H, Fang YQ et al. Clopidogrel reduces the inflammatory response of lung in a rat model of decompression sickness. Respir Physiol Neurobiol 2015; 211 : 9–16. doi: 10.1016/ j.resp.2015.02.003.
33. Umemura K, Ishihara H, Nakashima M. Anti-platelet effects of clopidogrel in rat middle cerebral artery thrombosis model. Thromb Res 1995; 80(3): 209–216.
34. Cortelekoglu T, Bozkurt AK, Ustundag N et al. The effects of clopidogrel and calcium dobesilate on intimal hyperplasia following vascular injury. Acta Chir Belg 2006; 106(2): 206–210.
35. Graff J, Harder S, Wahl O et al. Anti-inflammatory effects of clopidogrel intake in renal transplant patients: effects on platelet-leukocyte interactions, platelet CD40 ligand expression, and proinflammatory biomarkers. Clin Pharmacol Ther 2005; 78(5): 468–476. doi: 10.1016/ j.clpt.2005.08.002.
36. Herbert JM, Bernat A, Maffrand JP. Importance of platelets in experimental venous thrombosis in the rat. Blood 1992; 80(9): 2281–2286.
37. Joseph B, Pandit V, Aziz H et al. Clinical outcomes in traumatic brain injury patients on preinjury clopidogrel: a prospective analysis. J Trauma Acute Care Surg 2014; 76(3): 817–820. doi: 10.1097/ TA.0b013e3182aafcf0.
38. Peck KA, Calvo RY, Schechter MS et al. The impact of preinjury anticoagulants and prescription antiplatelet agents on outcomes in older patients with traumatic brain injury. J Trauma Acute Care Surg 2014; 76(2): 431–436. doi: 10.1097/ TA.0000000000000107.
39. Ivascu FA, Howells GA, Junn FS et al. Predictors of mortality in trauma patients with intracranial hemorrhage on preinjury aspirin or clopidogrel. J Trauma 2008; 65(4): 785–788. doi: 10.1097/ TA.0b013e3181848caa.
40. Zhang X, Chen Y, Jenkins LW et al. Bench-to-bedside review: apoptosis/ programmed cell death triggered by traumatic brain injury. Crit Care 2005; 9(1): 66–75. doi: 10.1186/ cc2950.
41. Liou AK, Clark RS, Henshall DC et al. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol 2003; 69(2): 103–142.
42. Jenkins LW, Moszynski K, Lyeth BG et al. Increased vulnerability of the mildly traumatized rat brain to cerebral ischemia: the use of controlled secondary ischemia as a research tool to identify common or different mechanisms contributing to mechanical and ischemic brain injury. Brain Res 1989; 477(1–2): 211–224.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2019 Číslo 5
Nejčtenější v tomto čísle
- Treatment of insomnia in the context of neuropathic pain
- Compressive neuropathies as an occupational disease
- Changes of paraspinal muscle morphology in patients with chronic non-specific low back pain
- Endoscopic surgery for lumbar disc herniation – the first experience