
Neurologické poruchy v rámci kritického stavu
Neurological Disorders in Critical Illness
Critical illness is usu ally defined as a conditi on characterised by failure of one or more organs or systems as a result of a seri o us dise ase or tra uma and usu ally accompani ed by systemic inflammatory response syndrome. Among ca uses of critical illness are infecti ons, usu ally respiratory ones, tra umas, burns, gre at surgery or primary failure of one organ including central nervo us system or ne uromuscular failure. Original concept of sepsis as an inappropri ate mobilizati on of defence mechanisms against virulent infecti on was modifi ed by a concept of systemic inflammatory response syndrome as uncontrolled wide inflammatory response against not only infecti on and le ading into multiple organ failure. Failure of cerebral functi ons in the form of diffuse encephalopathy – encephalopathy in critical illness – is usu ally an integral part of multiple organ failure. Among key signs are decre ased level of consci o usness and disordered cognitive functi ons (especi ally attenti on, cogniti on and ori entati on). Most scoring systems of multiple organ failure use the Glasgow coma scale to score the central nervo us system failure. The aeti ology of encephalopathy in critical illness is probably multi - factori al and overlaps with that of eti ologically unspecific syndrome of deliri um. Encephalopathy in critical illness sho uld be differenti ated from infecti o us encephalitis and acute para - infecti o us a uto immune encephalopathi es. Ne uromuscular disorders in critically ill pati ents demonstrate especi ally as a new we akness (“critical illness we akness”). Beside exacerbati on of pre - existing ne uromuscular disorder and persistent pharmacological blockade of ne uromuscular transmissi on ca used by non‑depolarising muscle blocking agents, there exists the newly recognised ca use called polyne uropathy and myopathy of critical illness. These disorders affect to some extend at le ast half of critically ill pati ents. Aeti ology is probably multi - factori al. Systemic inflammatory response syndrome and multiple organ failure are probably among important eti ological factors. In critical illness myopathy, “functi onal denervati on” ca used by non‑depolarising muscle blocking agents and high doses of corticostero ids have additi onal effect. Both conditi ons are frequently associ ated in the same pati ent – critical illness polyne uromyopathy. In contrast to original concept of complicati ons of critical illness these conditi ons seem to be an integral part of multiple organ failure – a ne uromuscular failure – analogical to the failure of other organs and systems. Critical illness polyne uropathy and myopathy are important risk factors of prolonged morbidity and mortality in critically ill pati ents. Current state of art in the are a of aeti ology and pathogenesis of critical illness doesn’t enable the use of effective preventi on and tre atment of ne uromuscular disorders.
Key words:
critical illness – sepsis – multiple organ failure – encephalopathy – polyne uropathy – myopathy
Autoři:
prof. MUDr. Josef Bednařík, CSc.
Působiště autorů:
LF MU a FN Brno
; Ne urologická klinika
Vyšlo v časopise:
Cesk Slov Neurol N 2008; 71/104(5): 511-529
Kategorie:
Minimonografie
Poděkování: a utor děkuje prof. MUDr. Zdeňku Lukášovi, CSc., za poskytnutí histopatologických nálezů.
Souhrn
Kritický stav je obvykle definován jako stav spojený se selháním jednoho nebo více orgánových systémů, který vznikl na podkladě závažného onemocnění nebo úrazu. Jeho nedílno u so učástí je syndrom systémové zánětlivé odpovědi, který je závažným onemocněním nebo úrazem spo uštěn. Příčino u rozvoje kritického stavu je nejčastěji infekce, obvykle respirační, dále poranění, popálení, velká operace či primární selhání jednoho orgánu či systému včetně primárního postižení centrálního nervového systému nebo ne uromuskulárního systému. Původní koncepce sepse jako ne adekvátní mobilizace obranných mechanizmů proti virulentní infekci byla modifikována konceptem syndromu systémové zánětlivé odpovědi. Jedná se o nekontrolované široké zánětlivé re akce nejen na infekci, jež vedo u až k multi orgánovému selhání. So učástí multi orgánového selhání může být selhání mozkových funkcí charakteru difuzní encefalopati e – tzv. encefalopati e kritického stavu. Manifestuje se kombinací kvantitativní a kvalitativní poruchy vědomí (zejména poruchy pozornosti, vnímání, ori entace a dalších kognitivních funkcí). Většina systémů skórujících multi orgánové selhání po užívá ke kvantifikaci selhání centrálního nervového systému Glasgovsko u škálu bezvědomí. Eti ologi e encefalopati e kritického stavu je zřejmě multifaktori ální a překrývá se s eti ologicky nespecifickým syndromem deliri a. Diferenci álně di agnosticky je třeba odlišit zejména infekční encefalitidy a akutní parainfekční a uto imunitní encefalopati e. Ne uromuskulární poruchy u kriticky nemocných se manifestují zejména nově vzniklo u svalovo u slabostí. Kromě exacerbace preexistujícího nervosvalového onemocnění a perzistující farmakologické blokády nervosvalového přenosu navozené působením nedepolarizujících blokátorů nervosvalového přenosu je nově rozpoznano u příčino u tzv. polyne uropati e a myopati e kritického stavu. Tyto poruchy postihují v různém stupni minimálně polovinu kriticky nemocných. Eti opatogeneze je pravděpodobně multifaktori ální. Významným eti ologickým faktorem je syndrom systémové zánětlivé odpovědi a multi orgánové selhání. U myopati e kritického stavu je pravděpodobným přídatným faktorem „funkční denervace“ navozená blokátory nervosvalového přenosu a vysoké dávky kortikostero idů. Obě jednotky se u jednotlivých nemocných velmi často kombinují – polyne uromyopati e kritického stavu. Oproti původní představě označující tyto poruchy jako komplikace kritického stavu se jedná spíše o so učást multi orgánového selhání – „nervosvalové selhání“ – analogické selhání dalších orgánů v rámci kritického stavu. Jso u významným rizikovým faktorem prolongované mortality a morbidity kriticky nemocných. Dosavadní stav poznatků o eti opatogenezi kritického stavu ne umožňuje účinno u prevenci či léčbu nervosvalových poruch.
Klíčová slova:
kritický stav – sepse – multi orgánové selhání – encefalopati e – polyne uropati e – myopati e
I. Sepse, kritický stav, multi orgánové selhání
V so uvislosti se zlepšováním intenzivní péče narůstá počet nemocných, kteří přežívají dříve smrtelné onemocnění či poranění, často však za cenu rozvoje tzv. kritického stavu. Kritický stav (Critical Illness) je obvykle definován jako stav spojený se selháním jednoho nebo více orgánových systémů, který vznikl na podkladě závažného onemocnění nebo úrazu. Jeho nedílno u so učástí je syndrom systémové zánětlivé odpovědi, který je závažným onemocněním nebo úrazem spo uštěn.
Synonymem kritického stavu je těžká sepse či septický šok (zejména pokud dominuje selhání oběhu). So učasno u dysfunkci či selhání více orgánů nebo systémů označujeme jako multi orgánovo u dysfunkci (Multiple Organ Dysfuncti on, MOD) či multi orgánové selhání (Multiple Organ Failure, MOF).
Sepse (také septický syndrom) byla původně považována za nutno u, ale ne adekvátní mobilizaci obranných mechanizmů proti virulentní infekci [1]. Změnu náhledu na tuto problematiku způsobila dvě zásadní pozorování. Prvním bylo zjištění [2], že medi átory získané z aktivovaných le ukocytů moho u vyvolat široko u zánětlivo u odpověď (tzv. syndrom systémové zánětlivé – v české literatuře také zánětové – odpovědi, Systemic Inflammatory Response Syndrome, SIRS) a MOF. Bone et al [3] pak zjistili, že široká zánětlivá re akce vedo ucí k multi orgánovému selhání se může objevit nejen jako následek infekce, ale také tra umatu nebo krvácení bez přítomnosti infekce. Vznikl koncept SIRS jako nekontrolované široké zánětlivé re akce vedo ucí až k šoku a multi orgánovému selhání. Termín sepse se připo uští pro označení zánětlivé systémové odpovědi infekčního původu, což je asi 60 % případů.
Sepse byla považována za příklad nekontrolované aktivace prozánětlivých mechanizmů. V so učasné době se však objevují názory, že může jít o dysregulaci tlumících, dezaktivačních mechanizmů zánětu, což vede k přetrvávající prozánětlivé re akci. U kritického stavu může být časná prozánětlivá odpověď vystřídána protizánětlivo u odpovědí. Časový vztah mezi pro - a protizánětlivým stavem však nebyl plně objasněn a mnohdy nemocný několikrát osciluje mezi oběma extrémy. Následně byl popsán syndrom analogický SIRS – syndrom kompenzační protizánětlivé odpovědi (Compensatory Anti‑Inflammatory Response Syndrome, CARS) provázený zvýšeno u produkcí protizánětlivých cytokinů [4]. Interakce mezi pro - a protizánětlivými medi átory určuje tíži imunitní dysfunkce. Buďto je dosaženo rovnováhy, nebo převáží pro - či protizánětlivé medi átory vedo ucí k SIRS či CARS a ústící v MOD či MOF [1]. Zvýšená hladina některých prozánětlivých (interle ukiny 1 a 6 – IL‑1, IL‑6, Tumor Necrosis Factor alpha, TNF‑a) – i protizánětlivých cytokinů (IL‑10) byla spojena s nepříznivo u prognózo u kritického stavu [5 – 6].
Příčino u rozvoje kritického stavu je často infekce, nejčastěji respirační, dále poranění, popálení, velká operace či primární selhání jednoho orgánu či systému. Může jít rovněž o primární onemocnění či poranění nervového systému, nejčastěji mozku (např. cévní mozková příhoda, meningoencefalitida či krani ocerebrální tra uma), ale také primární ne uromuskulární onemocnění, např. Guillainův - Barrého syndrom, myastheni a gravis či amyotrofická laterální skleróza. Zejména jde o stavy, které lokalizací a tíží postižení vedo u k ventilačnímu či kardi ovaskulárnímu selhání.
So učástí kritického stavu je so učasná či postupná dysfunkce až selhání základních orgánů a systémů. K definici dysfunkce a selhání jednotlivých systémů a kvantifikace tíže multi orgánové dysfunkce a selhání se po užívá řada skórovacích systémů. Mezi nejčastěji užívané skórovací systémy patří Acute Physi ology and Chronic He alth Evalu ati on (APACHE) verze II [7] a III [8] a jednodušší skóre navržené evropsko u expertní skupino u: the Sequenti al Organ Failure Assessment score (SOFA), tab. 1, [9 – 11]. SOFA či APACHE II a III korelují s prognózo u kriticky nemocných vyjádřeno u délko u pobytu na jednotce intenzivní péče či mortalito u [10 – 12].

Oba systémy po užívají ke skórování ne urologické dysfunkce či selhání Glasgovsko u škálu bezvědomí (The Glasgow Coma Scale, GCS). U nemocných v kritickém stavu se ovšem vyskytuje ve vysokém procentu postižení nejen mozku, ale i míchy, periferního nervového systému a kosterního svalstva. Dysfunkce a selhání mozku jso u považovány za so učást MOF, zatímco nervosvalové poruchy jso u označovány jako komplikace kritického stavu a jejich projevy nejso u so učástí skórovacích systémů (i když moho u ovlivnit např. GCS). Dle so učasných poznatků jde však i v případě postižení periferního nervového systému či kosterního svalstva spíše o dysfunkci nebo selhání dalšího systému v rámci MOF [13].
Ne urologické poruchy v rámci kritického stavu lze rozdělit na poměrně časné postižení mozku (tzv. encefalopatii kritického stavu), míchy (vzácno u hypoxicko u myelopatii) a ne uromuskulární poruchy [14].
II. Encefalopati e kritického stavu (CIE)
Již Hippokrates a Galen zaznamenali, že deliri um (podle Hippokrata „phrenitis“) může doprovázet zánět [15]. Jde o nejčastější encefalopatii, se ktero u se setkáme v intenzivní péči, podle odhadů postihuje 23 – 70 % nemocných se sepsí [16 – 18]. Terminologi e je nejednotná, nejčastěji se po užívá termín septická encefalopati e (SE), který by však měl být vyhrazen po uze pro stavy spojené se sepsí. Nejběžnější klinicko u manifestací je syndrom deliri a, proto se mluví o deliri u na jednotce intenzivní péče (Intensive Care Unit Deliri um). Zřejmě nejvhodnějším popisným termínem vyjadřujícím multifaktori ání eti ologii encefalopati e je encefalopatie klitického stavu (Encephalopathy in Critical Illness, CIE).
Klinická manifestace
CIE se manifestuje obvykle do 14 dnů od vzniku kritického stavu. Klinická manifestace odpovídá difuzní encefalopatii a zahrnuje poruchy kognitivních funkcí (poruchy pozornosti, paměti, ori entace), rozvinutý syndrom deliri a i kvantitativní poruchu vědomí včetně kómatu. So učasně jso u febrili e, laboratorní známky zánětu a obvykle i známky dysfunkce či selhávání více orgánů. Nevyskytují se obvykle meninge ální příznaky, ložiskové příznaky, epileptické záchvaty ani motorické symptomy typické pro metabolické encefalopati e (asterixis, tremor, multifokální myoklonus).
Di agnostika
CIE je ne zcela jasně definovaná jednotka, která doprovází rozvoj kritického stavu. Jde o di agnózu per exclusi onem, protože neexistuje žádný specifický di agnostický test. Diferenci álně di agnosticky je nutné odlišit encefalopati e vzniklé v důsledku primární izolované dysfunkce či selhání kardi ovaskulárního, respiračního, jater či ledvin, které předchází rozvoji sepse a multi orgánového selhání. V praxi je však často obtížné rozlišit, zda je encefalopati e důsledek orgánového selhání, nebo následek sepse a so učást multi orgánového selhání.
Druho u skupino u encefalopati í, ktero u je třeba zejména od SE diferencovat, je akutní infekční encefalitida (způsobená přímo u invazí infekčního agens do mozku), akutní zánětlivá parainfekční encefalopati e předpokládané a uto imunitní patogeneze s dvěma patologicky dobře definovanými formami – akutní diseminovano u encefalomyelitido u(ADEM) a akutní hemoragicko u le ukoencefalopati í (AHLE) – také zvano uHurstova choroba, a nepřesně definované akutní toxické encefalopati e vyskytující se ve věku do dvo u let a vedo ucí k difuznímu mozkového edému [19] (např. Reyův syndrom), (tab. 2). Základem rozlišení je jednak klinický obraz, jednak zejména vyšetření mozkomíšního moku, které je u SE normální nebo vykazuje lehko u hyperproteinorachii, zatímco u encefalitidy i parainfekčních encefalopati í je přítomna pleocytóza (s výjimko u akutních toxických encefalopati í), hyperproteinorachi e a u encefalitidy lze detekovat přítomnost mikrobů.
![Diferenciální diagnostika encefalopatií asociovaných s infekcí [19].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/721172aba4a84745079ded7503966fc5.png)
EEG abnormita je sice citlivým indikátorem encefalopati e kritického stavu, abnormální EEG se však nachází i u dalších výše zmíněných encefalopati í; stupeň abnormity koreluje u SE s tíží klinického průběhu [20]. Obdobná charakteristika platí pro abnormitu korových komponent somatosenzorických evokovaných potenci álů [21].
Zobrazovací nálezy v oblasti mozku jso u u CIE bez pozoruhodností na rozdíl od encefalitid i parainfekčních encefalopati í.
Di agnostika CIE je velmi obtížná u primárních ložiskových lézí mozku, které jso u příčino u kritického stavu, např. u akutních cévních mozkových příhod, kde lze epizodu deliri a charakterizovano u klinicky náhlo u změno u stavu a následno u fluktu ací zachytit až u 45 % nemocných [22,23].
Eti opatogeneze
Je zřejmě multifaktori ální. U animálního modelu septické encefalopati e dochází k poruše hematoencefalické bari éry a perivaskulárnímu edému [24]. Zvažuje se efekt medi átorů zánětu (TNF‑a, interferony alfa, gamma, interle ukin‑1 beta), které mají vliv na funkci ne uronů i hematoencefalické bari éry [24 – 27]. Změny endoteli ální perme ability spolu s dalšími mikrovaskulárními cerebrálními změnami by mohly vést k zvýšenému průniku „falešných ne urotransmiterů“ (např. některých aminokyselin: fenylalaninu, tryptofanu) do mozku. Metabolit tryptofanu – kyselina chinolinová může aktivovat receptor N - metyl - D - aspartátu (NMDA), aktivovat ne uronální izoformu NO - syntetázy, která spolu s dalšími volnými radikály může poškodit DNA a aktivovat jaderný enzym poly - ADP - riboso - syntetázu (PARS), což může vést k energetické depleci a buněčné smrti. Dalším zvažovaným faktorem u CIE je těžká arteri ální hypotenze [16]. U paci entů, kteří zemřeli na septický šok, jso u přítomny ischemické změny v mozkovém kortexu. U animálního modelu septické encefalopati e bez oběhového selhání nalézáme degenerativní změny ne uronů frontálního kortexu, není však známo, zda obdobné změny jso u přítomny i u člověka [28].
Klinická manifestace CIE se překrývá se syndromem deliri a, který se vyskytuje nejen u sepse a nemocných v kritickém stavu, ale jako eti ologicky nespecifický syndrom je přítomen prakticky u všech závažnějších cerebrálních i extracerebrálních onemocnění a stavů. Je tedy otázko u, zda patogeneze CIE je odlišná od patogeneze deliri a obecně [22].
Mezi rizikové faktory deliri a detekovatelné v séru patří např. zvýšení poměru ure a/ kre atinin [29], abnormální hypo - či hyperkalemi e, natremi e a glykemi e [30]. Významným rizikovým faktorem deliri a je preexistující demence [31,32]. Lze tedy předpokládat, že genetické rizikové faktory demence moho u být so učasně i rizikovými faktory deliri a. Mezi takové potenci álně rizikové faktory patří alela apolipoproteinu E4 asoci ovaná s časným rozvojem Alzheimerovy choroby nebo alela A9 dopaminergního transportního systému asoci ovaná s rozvojem deliri a tremens u přerušení abúzu alkoholu, vztah k deliri u však dosud nebyl potvrzen [33].
Patogeneze deliri a stejně jako CIE není dosud uspokojivě vysvětlena. Příčino u může být velká heterogenita jak syndromu deliri a, tak studovaných populací paci entů. I když může deliri um vznikno ut v důsledku anatomické mozkové léze, jako je cévní mozková příhoda, ve většině případů vzniká dysbalancí klíčových centrálních ne urotransmiterů, zejména sníženo u aktivito u cholinergního a zvýšeno u aktivito u serotoninergního systému [34 – 36]. Deliri um se vyskytuje u řady stavů, kde je přítomen zánět: zejména jde o infekce, po operační a posttra umatické stavy s rozvojem SIRS. Zánět vede k poruše hematoencefalické bari éry a snižuje cholinergní transmisi. Medi átory zánětu, zejména cytokiny, tak moho u být významnými bi omarkery deliri a. Existují studi e prokazující zvýšeno u hladinu těchto markerů u paci entů s deliri em [37].
Prognóza
Je obtížné ji poso udit nezávisle od prognózy kritického stavu, která je sama od sebe závažná.
Léčba a prevence
Základem je komplexní léčba základního onemocnění ruku v ruce s důsledně prováděno u podpůrno u intenzivní péčí, v rámci které je třeba léčit sepsi, zabezpečit dostatečno u perfuzi mozku a zabránit mozkové hypoxii. Intenzivně je zko umán efekt různých antagonistů zánětlivých medi átorů. Dalšími terape utickými možnostmi, jejichž efekt je třeba prokázat, je aplikace antagonistů glutamátových receptorů, zametačů volných radikálů nebo inhibitorů NO a PARS.
III. Hypoxická myelopati e
Jde o raritní jednotku, navíc se sporným vztahem ke kritickému stavu, sepsi a multi orgánovému selhání [38]. Poněkud lépe definovaný, i když rovněž raritní je tzv. Hopkinsův syndrom, také označovaný jako akutní post‑astmatická amyotrofi e [39]. Jde o akutní ataku svalové slabosti po těžkém astmatickém záchvatu, která připomíná poli omyelitidu. Většina případů byla popsána u dětí do 13 let věku. Jde o chabo u parézu jedné nebo dvo u končetin v důsledku předpokládaného postižení předních rohů míšních. Kromě hypoxi e je zvažována i virová eti ologi e.
IV. Ne uromuskulární poruchy u kriticky nemocných
Velmi častým klinickým příznakem u kriticky nemocných je nově vzniklá svalová slabost (tzv. slabost kritického stavu, Critical Illness We akness). Na rozdíl od centrálních paréz v důsledku primární mozkové léze – příčiny kritického stavu (např. akutní cévní mozkové příhody) – je svalová slabost vzniklá v průběhu kritického stavu obvykle ne uromuskulárního původu. Je jediným (myopati e, blokáda nervosvalového přenosu) nebo dominujícím příznakem (polyne uropati e) ne uromuskulárního postižení. U řady ventilovaných nemocných může být prvním a v některých případech i jediným příznakem ne uromuskulární poruchy slabost respiračního svalstva. Projevuje se ztíženým odpojením od ventilátoru v době, kdy pominuly jiné důvody umělé plicní ventilace. Nově vzniklé parézy je však obtížné objektivizovat, neboť kriticky nemocní obvykle nespolupracují. Jso u často v bezvědomí či tlumeni sedativy či opi o idy, je přítomno paretické postižení centrálního původu v důsledku primárního postižení mozku nebo míchy či farmakologické blokády nervosvalového přechodu. Všechny tyto faktory způsobují, že asi v polovině případů postižení typu myopati e či polyne uropati e nejso u přítomny spolehlivé klinické příznaky, které by umožnily tyto ne uromuskulární komplikace di agnostikovat, a je nutná di agnostika elektrofyzi ologická či histopatologická (tab. 3).
Příčiny získané slabosti u kriticky nemocných ne uromuskulárního původu je možno rozdělit do tří skupin:
A. Exacerbace preexistujícího nervosvalového onemocnění v důsledku rozvoje kritického stavu a multi orgánového selhání; mezi nejčastější nervosvalová onemocnění dekompenzující se u kriticky nemocného patří:
- myastheni a gravis
- svalová dystrofi e
- amyotrofická laterální skleróza
- peri odické paralýzy
- akutní intermitentní porfyri e
- Guillainův - Barrého syndrom (GBS). Tenpředstavuje asi nejvýznamnější diferenci álně di agnostický problém: častěji jde o primární onemocnění, které může být komplikováno rozvojem kritického stavu, může však jít i o pozdní komplikaci u paci enta v kritickém stavu. V diferenci ální di agnostice oproti samostatným ne uromuskulárním poruchám kritického stavu pomáhá elektrofyzi ologický obraz demyelinizační ne uropati e u většiny případů GBS a dále proteinocytologická disoci ace [40 – 41].
B. Perzistující blokáda nervosvalového přenosu v důsledku protrahovaného působení nedepolarizujících blokátorů nervosvalového přenosu (NBNP). Blokátory nervosvalového přenosu jso u léky vyvinuté původně pro krátkodobo u aplikaci na operačním sále a po užívané u asistované ventilace. U kriticky nemocných dochází často k nežádo ucímu protrahovanému efektu NBNP [42]. Příčino u je změněná farmakodynamika a farmakokinetika ve vyšším věku (značná část kriticky nemocných má vyšší věk) či v důsledku selhání některých orgánů, zejména jater a ledvin. Přetrvávající blokádu nervosvalového přenosu lze přitom velmi obtížné detekovat klinicky. Doporučuje se dávku NBNP kontrolovat pomocí jednoduché elektrické stimulace motorických vláken n. ulnaris či n. medi anus séri í čtyř stimulů o frekvenci 2 Hz (tzv. train of fo ur) a sledování motorické kontrakce aspekcí či pomocí mechanogramu. Vymizení poslední odpovědi odpovídá blokádě přibližně 75 %, ztráta 3. odpovědi blokádě 80 %, chybění 2. odpovědi blokádě 90 % a chybění všech odpovědí blokádě 100 % nervosvalových plotének [43]. Citlivější detekce přetrvávající blokády nervosvalového přenosu je možná pomocí repetitivní elektrické stimulace motorického nervu a registrací dekrementu amplitudy či arey sumačního svalového akčního potenci álu. Snížit celkovo u dávku NBNP, a tím i minimalizovat riziko perzistující blokády nervosvalového přenosu, event. rozvoj myopati e kritického stavu moho u systémy a utomatického dávkování NBNP na základě monitorování úrovně svalové relaxace a jejího udržení na potřebné úrovni, řízené na principu fuzzy logiky [44].
C. Nově vzniklé ne uromuskulární poruchy kritického stavu
Ne uromuskulární poruchy v rámci kritického stavu v užším slova smyslu je možno rozdělit na dvě původně samostatné jednotky [40, 45]:
- polyne uropatii kritického stavu (Critical Illness Polyne uropathy, CIP) a
- myopatii kritického stavu (Critical Illness Myopathy, CIM).
- V poslední době narůstají důkazy o převažujícím kombinovaném postižení typu polyne uromyopati e kritického stavu (Critical Illness Polyne uromyopathy, CIMP).
ad 1) Polyne uropati e kritického stavu
Histori e
První zmínky o polyne uropatii u nemocných v kómatu jso u z roku 1961 [46] a u popálených z roku 1971 [47]. Polyne uropati e kritického stavu jako samostatná jednotka byla poprvé popsána v roce 1983 nezávisle na sobě dvěma skupinami [48,49]. V dalších letech byla upřesněna klinická, elektrofyzi ologická a histopatologická charakteristika této jednotky [14,50 – 54]. V naší literatuře byla tato problematika poprvé popsána v roce 2000 [55].
Epidemi ologi e
SIRS je přítomen u 20 – 50 % nemocných na jednotkách intenzivní péče [56] a u přibližně 70 % z nich se rozvine CIP [53,54]. Je však pravděpodobné, že výskyt polyne uropati e byl nadhodnocen, neboť u části případů šlo o myopatii kriticky nemocných. CIP je vzácně popisována i u pedi atrických paci entů [57,58].
Eti opatogeneze
Přesná příčina CIP není známa. Retrospektivní i prospektivní studi e až na výjimky ne uspěly ve snaze identifikovat některý z řady potenci álních eti ologických faktorů [59,60]. Některá ojedinělá sdělení i rozsáhlejší studi e prokazovaly sice asoci aci určitých faktorů s rozvojem CIP, zejména hypalbuminemi e, hyperglykemi e [53,59], hyperpyrexi e [60] či NBNP [42], v posledních letech však převládají názory, které spojují CIP se SIRS a multi orgánovým selháním [53,54]. Zvýšená hladina glukózy a snížená hladina albuminu v séru korelují sice s poklesem funkce periferního nervstva, obě bi ochemické změny jso u však dobře známým projevem sepse a multi orgánového selhání.
Nejnovější výzkumy umožňují spekulativní výklad vzniku CIP v rámci SIRS. U sepse je porušena mikrocirkulace v řadě orgánů [61]. Krevní cévy zásobující nervy postrádají schopnost a utoregulace [62], což je činí velmi náchylné k poškození. Cytokiny, které jso u produkované při sepsi, mají histaminové účinky a moho u zvyšovat mikrovaskulární perme abilitu. Výsledný endone urální edém může indukovat hypoxii uzávěrem kapilár či zvýšením interkapilární vzdálenosti. Hund et al [63] popsali v séru nemocných s CIP endogenní toxin, jehož podstata však není známa.
Zvýšená pozornost při výzkumu vzniku kritického stavu a jeho komplikací je věnována v poslední době i apoptóze. Na rozdíl od nekrózy je apoptóza regulovaný nezánětlivý proces, závislý na energii. Apoptóza je kritická zejména pro kontrolu le ukocytů a lymfocytů, které jso u v nadměrném množství aktivovány a produkovány jako re akce na infekci a které musejí být eliminovány, jakmile je infekce odstraněna. Byly pozorovány změny v apoptóze jak ne utrofilů, která je opožděná, tak lymfocytů, která je zrychlená. Význam těchto změn však není dosud objasněn [1].
Klinická manifestace
CIP se typicky projeví jako končetinová slabost a areflexi e, v těžkých případech dochází ke kvadruplegii a respirační paralýze, která se může manifestovat obtížným odvykáním od umělé plicní ventilace [63 – 65]. Slabost se obvykle manifestuje v průběhu 3. – 4. týdne trvání kritického stavu. Svalové atrofi e jso u pozdním a nespecifickým příznakem. V rámci polyne uropati e moho u být postižena i vlákna senzitivní a a utonomní, poruchu citlivosti a a utonomní dysfunkci lze však v podmínkách kritického stavu a jednotky intenzivní péče obtížně detekovat. U poloviny nemocných však klinické příznaky CIP nejso u patrné a jso u přítomny po uze elektrofyzi ologické známky akutní axonální polyne uropati e.
Di agnostika a diferenci ální di agnostika
Relativní podíl myopati e a ne uropati e u jednotlivého nemocného s novo u svalovo u slabostí je obtížné poso udit po uhým klinickým vyšetřením.
Elektrofyzi ologická vyšetření představují důležitý klíč k diferenci aci obo u jednotek, resp. poso uzení přítomnosti ne uropatické a myopatické komponenty, event. nově rozpoznané poruchy svalové excitability. Při standardním elektromyografickém vyšetření je možno diferencovat tři vzorce elektrofyzi ologického postižení [66]: čistě motorické postižení (snížení amplitudy sumačního svalového akčního potenci álu [CMAP], event. abnormální spontánní aktivita), které může být podmíněné distální axonální motoricko u ne uropati í, myopati í i porucho u nervosvalového přenosu, dále smíšeným motoricko‑senzitivním postižením, typickým pro část nemocných s polyne uropati í, a vzácně čistě senzitivním postižením (abnormální senzitivní ne urogram), svědčícím rovněž pro polyne uropatii. Prvním elektrofyzi ologickým projevem, který lze detekovat již během prvního týdne trvání kritického stavu, je pokles amplitudy CMAP.
V úvodu každého elektrofyzi ologického vyšetření u kriticky nemocného, který dostává či v posledních několika dnech dostal NBNP, je nutné provést vyšetření repetitivní stimulace motorického nervu k vylo učení podílu perzistující farmakologické blokády nervosvalového přenosu. Přehlédnutí této blokády může vést k chybné interpretaci výsledků elektrofyzi ologického vyšetření, zejména snížení amplitud sumačního svalového akčního potenci álu (CMAP). Vzácně může jít o jino u příčinu abnormálního dekrementu amplitudy či arey CMAP než perzistující blokádu způsobeno u NBNP, zejména o exacerbaci myastheni a gravis, potencovano u případně so učasno u medikací aminoglykosidy.
I po vylo učení blokády nervosvalového přenosu není elektrofyzi ologické odlišení mezi CIM a CIP jednoznačné, protože elektrofyzi ologické rysy myopati e a polyne uropati e kritického stavu jso u velmi podobné (tab. 4).

Elektrofyzi ologické nálezy včetně abnormálního nervosvalového jitteru u CIP ukazují, že jde o primárně axonální distální převážně motoricko u ne uropatii [67]. U myopati e kritického stavu však může být amplituda CMAP snížena stejně jako u CIP. Příčino u je atrofi e svalových vláken, jejich ztráta v důsledku nekrózy, případně snížená dráždivost sarkolemy, jak bylo prokázáno pomocí tzv. přímé svalové stimulace. Stejně tak může být zejména u nekrotické myopati e přítomna spontánní aktivita charakteru fibrilačních potenci álů a pozitivních ostrých vln. U nekrózy svalových vláken může být abnormální spontánní aktivita vyvolána funkční dyskonexí ploténkové zóny od aktivní membrány či od depolarizace svalového vlákna jako důsledek membránové dysfunkce. Elektrofyzi ologické rysy CIM jso u tedy podobné jako u CIP. Diferenci ace těchto dvo u jednotek je možná pomocí některých dalších elektrofyzi ologických parametrů. CIM nepostihuje senzitivní nervová vlákna, zatímco u CIP jso u amplitudy senzitivního nervového akčního potenci álu (SNAP) sníženy. Je však obtížné získat spolehlivé senzitivní odpovědi v podmínkách jednotekintenzivní péče. Amplituda SNAP můžebýt i u myopati e falešně snížena v důsledku otoku končetiny. Přítomnost abnormálního senzitivního ne urogramu, která je obvykle a utomaticky interpretována jako známka CIP, navíc nevylučuje so učasno u myopatii. Obě jednotky dále produkují odchylný náborový vzorec, který je však obtížné či nemožné studovat u nespolupracujících či komatózních nemocných. Pokud je slabost těžšího stupně, volní aktivita může navíc zcela chybět. Rutinní elektrofyzi ologické vyšetření má tedy omezeno u cenu v diferenci aci CIP a CIM.
Novo u možností, jak diferencovat CIM od CIP, je metoda přímé svalové stimulace prostřednictvím jehlových elektrod umístěných přímo do svalu (obr. 1) [68 – 73]. Elektrická odpověď získaná přímo u stimulací svalu (the direct muscle CMAP, dmCMAP) může být porovnána s odpovědí získano u nervovo u stimulací (the nerve - evoked CMAP, neCMAP) (obr. 2 a – c). Je‑li redukce amplitudy CMAP zjištěná při běžné kondukční studii důsledkem ztráty excitability svalové membrány (tedy při postižení na úrovni svalu), pak tato vlákna neodpovídají jak na stimulaci nervu, tak svalu. Poměr nerv/ sval je přibližně roven 1. Pokud je snížení amplitudy CMAP důsledkem denervace, vede přímá elektrická stimulace svalu k vyšší amplitudě motorické odpovědi než stimulace nervu; poměr nerv/ sval je tedy typicky snížen pod 1, obvykle je menší než 0,5. Dosud však není jasné, jaký je vztah snížené excitability sarkolemy k dalším elektrofyzi ologickým, histopatologickým i klinickým projevům CIM.

![a–c. Záznam přímé svalové stimulace [73 s laskavým svolením Springer Science + Business Media]. Horní křivky představují záznam svalové odpovědi vyvolané přímou svalovou stimulací intramuskulárně jehlovou elektrodou (dmCMAP), dolní křivky představují záznam svalové odpovědi při stimulaci nervu povrchovou elektrodou (neCMAP).
Obr. 2a ukazuje normální dmCMAP, sníženou neCMAP a nízký poměr ne/dmCMAP (0,05) svědčící pro neurogenní lézi (< 0,5).](https://pl-master.mdcdn.cz/media/image/1c442f639346f3b5f0340551fab0bf00.jpg?version=1711876483)


CIP je spojena s normální či lehce zvýšeno u sérovo u hladino u kre atinkinázy (CK).
Patologická anatomi e
Bi opsi e senzitivního nervu (obvykle n. suralis či n. perone us superfici alis) prokazuje známky primární axonální degenerace s maximem distálně, bez známek zánětu. Svalová bi opsi e vykazuje v akutním stadi u známky denervace, projevující se disperzní atrofi í svalových vláken obo u typů, ve stadi u reinervace pak dochází k typovému seskupování svalových vláken (obr. 3).

Prognóza
Prognóza kriticky nemocných závisí na příčině kritického stavu, komorbiditách, věku a řadě dalších okolností, ale v průměru se na polyvalentně zaměřených pracovištích mortalita pohybuje mezi 20 – 30 %. U nemocných, kteří přežijí kritické onemocnění, je obvyklá po uze částečná úprava ne urologického deficitu [74]. Úprava může trvat několik měsíců a nemocní s velmi těžkým stupněm postižení nemusejí dosáhno ut plné úpravy funkce. Trvalý ne urologický deficit může vznikno u zejména v důsledku kompresivních monone uropati í [65].
Léčba
Nejso u žádné důkazy, že některý specifický způsob léčby septického syndromu, jako po užití inotropních léků, antibi otik, parenterální či enterální výživa, je jakýmkoli způsobem odpovědný za vznik polyne uropati e [53,54]. Veškerá opatření působící preventivně či léčebně u septického syndromu jso u jedinými so učasnými možnostmi, jak léčit CIP.
Předpokládaná úloha SIRS a prozánětlivých cytokinů v eti opatogenezi kritického stavu a multi orgánového selhání i CIP vyústila v klinické pokusy se zablokováním těchto prozánětlivých cytokinů v počátcích SIRS. Byly po užity monoklonální a utoprotilátky proti endotoxinu, TNF‑a či antagonista rekombinantního lidského IL‑1 receptoru, výsledky byly však negativní [75 – 77]. Snížená funkce makrofágů ve stadi u CARS vedla ke snaze léčit sepsi pomocí interfernonu gama s pozitivními výsledky [78]. Metaanalýza ukázala pokles mortality paci entů se septickým šokem léčených intravenózním imunoglobulinem, který však nebyl dosud potvrzen metodologicky kvalitní studi í [79].
ad 2) Myopati e kritického stavu
Histori e
V roce 1977 MacFarlane a Rosenthal [80] poprvé popsali kvadruplegii u astmatika, který byl léčen vysokými dávkami hydrokortizonu a nedepolarizujících blokátorů nervosvalového přenosu. Následovala řada dalších sdělení popisující obdobné případy, obvykle ve spojitosti s léčbo u kortikostero idy a NBNP [81 – 86] a označující tuto jednotku jako akutní hydrokortizonová myopati e [80,87], akutní kvadruplegická myopati e [83], nekrotizující myopati e [88 – 90], myopati e silných vláken (Thick Filament Myopathy) [91,92] či myopati e kritického stavu (Critical Illness Myopathy, CIM) [51,93,94]. Existuje však řada sdělení popisujících výskyt myopati e bez aplikace těchto látek [95,96].
Epidemi ologi e
Výskyt CIM není znám zejména pro nespolehlivo u diferenci aci oproti polyne uropatii kritického stavu. Je pravděpodobné, že některé údaje týkající se prevalence CIM byly zkresleny tím, že případy myopati e byly identifikovány jako polyne uropati e. Obecně studi e založené na klinice a klasické elektrofyzi ologii nadhodnocovaly výskyt CIP [63], zatímco při po užití histopatologických nálezů a novějších elektrofyzi ologických metod byl zjišťován výskyt CIM minimálně na úrovni CIP nebo i vyšší [55,71 – 74,97 – 99].
Eti opatogeneze
Existuje řada důkazů, že CIM je heterogenní onemocnění, ve které může vyústit řada patogenetických dějů [94,100]. Jedno u hypotézo u, opřeno u o řadu experimentálních důkazů, je so učasné myotoxické působení kortikostero idů a NBNP. Byl pozorován vzestup exprese stero idních receptorů v denervovaném svalu [101] a selektivní ztráta myosinových filament po injekci dexametazonu do denervovaného svalu u krys [102]. Glukokortiko idy aktivují také ATP - ubiquitin‑dependentní proteolytický systém, který může hrát roli v degradaci myosinu [103]. Další a utoři demonstrovali zvýšeno u expresi calpainu (kalci em aktivovaného enzymu, podílejícího se na degradaci proteinů) [104]. Na základě těchto nálezů byla postulována tzv. dvo ustupňová hypotéza (a Two Hit Hypothesis), podle které stero idy působí na terénu funkční denervace způsobené NBNP vedo ucí ke zvýšené expresi stero idních receptorů; imobilizace může být přídatným faktorem [105,106].
CIM byla popsána po podání metylprednizolonu [83,86,89,90], hydrokortizonu [80,87], prednizonu [86], betametazonu [107] a dexametazonu [82,107] a dále po podání NBNP pankuroni a, vekuroni a a atrakuri a [81,108]. Není známa minimální dávka, která vede k rozvoji CIM, ani relativní podíl kortikostero idů a NBNP. Někteří a utoři považují za rozhodující efekt kortikostero idů [86], jiní a utoři upřednostňují vliv NBNP [90].
Alternativní hypotéza podporovaná elektrofyzi ologickým nálezem inexcitability svalové membrány při přímé svalové stimulaci [69 – 71] předpokládá abnormální klidovo u depolarizaci sarkolemy v důsledku porušené regulace i ontových kanálů, opět nejspíše vlivem kortikostero idů či kombinace s NBNP.
Další hypotéza považuje CIM za katabolický proces v důsledku sepse a uvolnění prozánětlivých cytokinů, což vede k degradaci svalových proteinů [95, 109, 110] a odpovídá normálnímu nálezu EMG, normální hladině CK a histologicky atrofii vláken typu 2 (také kachektická myopati e) [94,99,100]. Existuje řada sdělení popisujících výskyt CIM u nemocných se sepsí či multi orgánovým selháním a bez expozice kortikostero idům či NBNP [92,96,99,104].
Klinický obraz
Onemocnění se manifestuje generalizovano u svalovo u slabostí, která se projeví obvykle ve stadi u vysazení NBNP a odpojování od ventilátoru. Slabost na rozdíl od většiny myopati í postihuje proximální i distální svaly či dominuje distální slabost [111]. Vzácně jso u postiženy okohybné a mimické svaly [86,108,112]. Tonus je snížený, postupně se rozvíjí svalové atrofi e, reflexy moho u být normální, snížené až atypicky nevýbavné. Citlivost je typicky neporušená, pokud není so učasné postižení charakteru polyne uropati e kritického stavu.
Di agnostika a diferenci ální di agnostika
Patologická anatomi e
Ve svalové bi opsii jso u popisovány tři základní typy patologických změn [95].
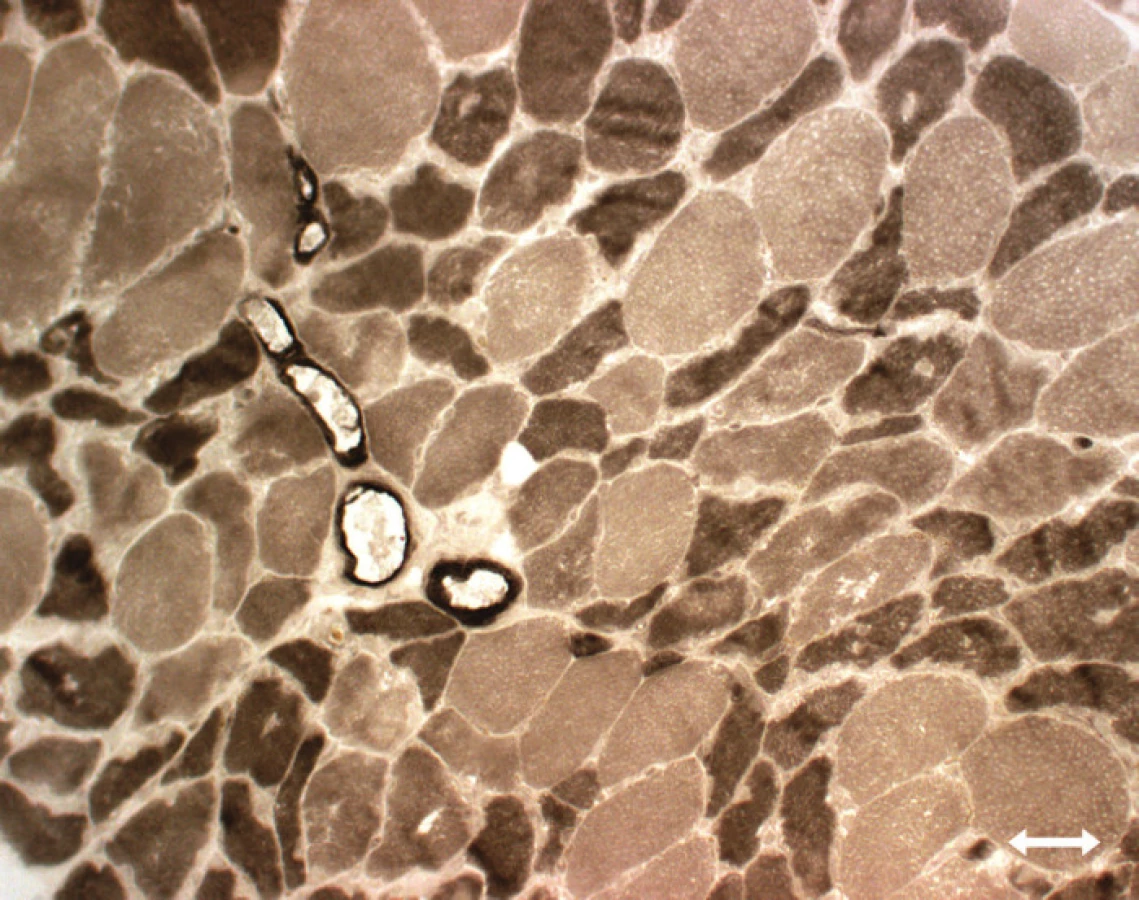
Atrofi e vláken typu 2 [89,95,113, 114]: tento typ patologi e je nespecifický, vyskytuje se při inaktivitě (Disuse Atrophy), u ne uropati e, u kriticky nemocných se kombinuje se ztráto u myosinu (obr. 4).
Nekróza svalových vláken, kolísající od mírné disperzní nekrózy až po těžko u panfascikulární nekrózu postihující vlákna obo u typů bez přítomnosti zánětlivého infiltrátu [83,88,112,114], či difuzní nekróza charakteru rabdomyolýzy se zvýšením CK a myoglobinuri í [88,90] – také akutní nekrotická myopati e (obr. 5) [89].

Za změnu, která je pro postižení svalové tkáně v rámci kritického stavu nejspecifičtější, je považována selektivní ztráta silných (myosinových) filament. Deficit myosinu je přítomen ve svalových vláknech obo u typů ložiskově či difuzně a je prokazatelný re akcí s myosinovo u ATP - ázo u, imunohistochemicky pomocí protilátek proti myosinu či pomocí elektronmikroskopického vyšetření (obr. 6) [83,86,89,91,94,102,106,113,114] – tzv. myopati e silných myosinových filament (Thick Filament Myopathy či Myosin Loss Myopathy). Tento typ patologi e je přítomen zejména u myopati e spojené s léčbo u kortikostero idy a NBNP. Není však zcela specifický u CIM a byl popsán u některých dalších myopati í, např. u dermatomyozitidy, kongenitálních myopati í či po transplantaci jater nebo plic [111,113,115].

V posledních letech se objevují sdělení prokazující bi optické nálezy svědčící pro podíl zánětlivých imunopatologických dějů na vzniku CIP. Jde o zvýšeno u expresi HLA I, membránového útočného komplexu (MAC), přítomnost makrofágů a T lymfocytů a produkci prozánětlivých a protizánětlivých cytokinů [97,116].
Kre atinkináza (CK) bývá pravidelně zvýšena u přibližně 50 – 85 % případů [82,117]. U nemocných se status asthmaticus léčených kortikostero idy v případě rozvoje CIM dosáhla hladina CK vrcholu 2. – 5. den od počátku expozice kortikostero idům a normalizovala se do 16 dnů [82]. Při výrazném zvýšení CK může být přítomna myoglobinuri e.
Elektrofyzi ologické vyšetření ukazuje typické známky myogenního postižení: snížení amplitudy CMAP, normální či lehce abnormální distální motorické latence a rychlosti vedení motorickým nervem a normální senzitivní ne urogram. V jehlové EMG jso u v případě zachování volní hybnosti přítomny potenci ály motorických jednotek o krátkém trvání a nízké amplitudě a dále rychlý až předčasný nábor motorických jednotek a případně spontánní aktivita typu fibrilačních potenci álů a pozitivních ostrých vln různé četnosti. Tento elektrofyzi ologický obraz je však velmi blízký nálezu u axonální polyne uropati e. Typické elektrofyzi ologické nálezy u obo u jednotek jso u shrnuty v tab. 2.
Při vyšetření přímo u svalovo u stimulací je u části paci entů s CIM a so učasno u sníženo u excitabilito u sarkolemy sval elektricky nedráždivý na rozdíl od CIP, u které je přítomna normální elektrická odpověď na přímo u svalovo u stimulaci a snížení amplitudy CMAP po stimulaci nervu [45,55,69 – 73].
Repetitivní stimulace motorického nervu je bez dekrementu, pokud není přítomna perzistující blokáda nervosvalového přenosu v důsledku předchozího podání NBNP.
Průběh a prognóza nejso u přesně známy. Pokud nemocní přežijí kritický stav, může být prognóza CIM dobrá [99].
Léčba: ka uzální léčba není známa. Obvykle se doporučuje preventivně omezit vliv potenci álně významných eti ologických činitelů, tj. omezit či vyvarovat se aplikace NBNP [118], minimalizovat dávky kortikostero idů, intenzivně léčit septický stav (antibi otika, podpora oběhu, zamezit malnutrici). Po odeznění kritického onemocnění by měla následovat časná mobilizace a rehabilitační péče.
ad 3) Polyne uromyopati e kritického stavu
Metodologická úskalí při rozlišení mezi polyne uropati í a myopati í kritického stavu a zejména často popisovaná ko incidence obo u typů postižení u týchž kriticky nemocných vedla k zavedení popisného termínu polyne uromyopati e kritického stavu [119,120] nebo myopati e a ne uropati e kritického stavu (CRIMYNE) [99,121]. Další po užívají termín paréza získaná na JIP (ICU - Acquired Paresis) a rezignují na rozlišení mezi myopati í a ne uropati í [59]. Řada novějších studi í po užívajících detailní elektrofyzi ologické a histopatologické metody prokazuje, že u kriticky nemocných jene uromuskulární systém postižen so učasně na úrovni periferních nervů i kosterních svalů, a to spíše multifokálně než difuzně [73,122 – 124].
Závěr
Přes velký pokrok v poznatcích o postižení nervového systému v rámci kritického stavu dosud existuje řada nevyřešených otázek v jejich di agnostice, eti opatogenezi, praktickém managementu, prevenci a léčbě:
- není známa přesná epidemi ologi e jakencefalopati e kritického stavu, tak ne uromuskulárních poruch, ale je jasné, že jde o mimořádně časté ne urologické postižení, jehož incidence bude v budo ucnu s postupující účinností intenzivní lékařské péče paradoxně narůstat.
- di agnostika CIE je založena na klinickém zhodnocení zejména ne uropsychi atrických příznaků a specifická elektro-di agnostická či zobrazovací di agnostika není známa. Neexistuje ani obecně přijímaný di agnostický algoritmus ne uropati e a myopati e kritického stavu. Klinická di agnostika je v podmínkáchkritického stavu nespolehlivá a málo senzitivní. Elektrofyzi ologická di agnostika je dostatečně senzitivní, ale ne - umožňuje zcela spolehlivě diferencovat postižení typu CIP a CIM. Svalová bi opsi e představuje nejspolehlivější průkaz myogenního postižení u kriticky nemocných a je so učástí navržené Lacomisovy klasifikace CIM [117], která však nebyla bez výhrad akceptována. Vzhledem k invazivnímu charakteru není svalová bi opsi e vyžadovaná v rutinní klinické di agnostice a doporučuje se obvykle po uze v případě, kdy je nutné odlišit jiné myopati e, zejména zánětlivé.
- není jasné, zda charakteristické patologické změny svalové tkáně v rámci CIM reprezentují samostatné jednotky, případně i s odlišno u eti opatogenezí, či jde o plynulé kontinuum změn včetně postižení periferního nervstva v rámci konceptu polyne uromyopati e kritického stavu.
- eti opatogeneze jak ne uromuskulárních poruch, tak zejména CIE není dosud přesvědčivě objasněna. Zatímco u polyne uropati e kritického stavu je přesvědčivě doložen eti ologický význam SIRS, u myopati e kritického stavu není i přes řadu experimentálních údajů a klinických sdělení přesvědčivě prokázaná úloha kortikostero idů a zejména NBNP v její eti opatogenezi a sílí názory ohledně významu SIRS a MOF. Nejpravděpodobnější je multifaktori ální eti ologi e, kdy jednotlivé faktory se navzájem podmiňují a na sebe navazují. Významnější podíl SIRS na eti opatogenezi CIM a její častá asoci ace s CIP stejně jako časný pokles amplitudy CMAP při elektrofyzi ologickém vyšetření by mohly napovídat, že jde o „ne uromuskulární selhání“, které je analogické k selhání dalších orgánů či systémů v rámci kritického onemocnění včetně selhání mozku, spíše než k pozdní „komplikaci“ kritického stavu.
- nedostatečné znalosti o příčinách a mechanizmech ne urologických poruch v rámci kritického stavu jso u důsledkem dosud velmi ne uspokojivé prevence a léčby těchto stavů. V so učasnosti se léčba zaměřuje na časné a účinné zvládnutí základního onemocnění a výzkum se zaměřuje na možnosti ovlivnění přestřelených a oscilujících imunopatologických pro - a protizánětlivých re akcí a apoptózy.
Po užité zkratky
ADEM akutní diseminovaná encefalomyelitida
AHLE akutní hemoragická le ukoencefalopati e
APACHE Acute Physi ology and Chronic He alth Evalu ati on
CARS syndrom kompenzační protizánětlivé odpovědi (Compensatory Anti‑inflammatory Response Syndrome)
CIE encefalopati e kritického stavu (Encephalopathy in Critical Illness)
CIM myopati e kritického stavu (Critical Illness Myopathy)
CIP polyne uropati e kritického stavu (Critical Illness Polyne uropathy)
CIPM polyne uromyopati e kritického stavu (Critical Illness Polyne uromyopathy)
CK kreatinkináza (Creatinkinase)
CMAP sumační svalový akční potenci ál (Compo und Muscle Acti on Potenti al)
CRIMYNE myopati e a ne uropati ekritického stavu (CRIticalIllness MYopathy and NEuropathy)
dmCMAP CMAP získaný přímo u stimulací svalu (the direct muscle CMAP)
GBS Guillainův - Barrého syndrom
GCS Glasgovská škála bezvědomí (the Glasgow Coma Scale)
IL interle ukin
MAC membránový útočný komplex (Membrane Attack Complex)
MOD multi orgánová dysfunkce (Multiple Organ Dysfuncti on)
MOF multi orgánové selhání (Multiple Organ Failure)
NBNP nedepolarizující blokátory nervosvalového přenosu
neCMAP CMAP získaný nervovo u stimulací (the nerve - evoked CMAP)
NMDA N - metyl - D - aspartát
PARS poly - ADP - riboso-syntetáza
SE septická encefalopati e
SIRS syndrom systémové zánětlivé odpovědi (Systemic Inflammatory Response Syndrome)
SNAP senzitivní nervový akční potenci ál (Sensory Nerve Acti on Potenti al)
SOFA the Sequenti al Organ Failure Assessment score
TNF‑α Tumor Necrosis Factor alpha
prof. MUDr. Josef Bednařík, CSc.
Ne urologická klinika
LF MU a FN Brno
Jihlavská 20
639 00 Brno
e‑mail: jbednar@fnbrno.cz
Přijato k recenzi: 22. 4. 2008
Přijato do tisku: 27. 5. 2008
Recenze:
prof. MUDr. Zdeněk Ambler, DrSc.
doc. MUDr. Edvard Ehler, CSc.
prof. MUDr. Pavel Ševčík, CSc.
prof. MUDr. Josef Bednařík, CSc.

Prof. MUDr. Josef Bednařík, CSc., vystudoval Lékařsko u fakultu Masarykovy univerzity v Brně. Od promoce v roce 1979 působil na 1. ne urologické klinice FN u sv. Anny v Brně a od roku 1993 až do so učasnosti pracuje na Ne urologické klinice FN Brno: od roku 1993 jako zástupce přednosty pro školství a od roku 2007 jako přednosta. V roce 1994 se habilitoval a v roce 2002 byl jmenován profesorem ne urologi e. Ve své klinické a vědecko‑výzkumné práci se věnuje zejména nervosvalovým a vertebrogenním onemocněním a klinické ne urofyzi ologii. Publikoval 70 původních prací in extenzo, z toho 25 v zahraničních impaktovaných časopisech, 30 přehledových prací, je a utorem nebo hlavním editorem několika monografi í a učebnic. Jeho práce byly 200krát citovány v Sci ence Citati on Index a byly odměněny řado u domácích cen, např. dvakrát Haškovcovo u ceno u. Prezentoval 300 přednášek a posterů (z toho 70 na mezinárodních kongresech). Je řešitelem a spoluřešitelem 10 obhájených grantů IGA MZČR. Je členem výboru České ne urologické společnosti nebo jeho revizní komise nepřetržitě od roku 1992. Je zakládajícím členem ne uromuskulární sekce ČLS JEP a členem jeho výboru od založení v roce 1991. Je členem redakce České a slovenské ne urologi e a ne urochirurgi e od roku 2000, zástupcem vedo ucího redaktora od roku 2002 a od roku 2005 působí jako její vedo ucí redaktor.
Vědomostní test
1. Kritický stav (Critical Illness) je definován jako:
- a) kardiorespirační selhání
- b) multiorgánové selhání
- c) kombinace multi orgánového selhání a SIRS
- d) synonymem je těžká sepse či septický šok
2. Syndrom systémové zánětlivé odpovědi (SIRS) je:
- a) synonymem sepse
- b) termín zahrnující oproti sepsi i systémovo u zánětlivo u odpověd neinfekčního původu
- c) považován za příklad nekontrolované aktivace prozánětlivých mechanizmů
- d) považován za hlavní eti opatogenetický faktor multi orgánového selhání
3. The Sequenti al Organ Failure Assessment score (SOFA):
- a) představuje skóre navržené evropsko u expertní skupino u ke kvantifikaci tíže multi orgánového selhání
- b) ke kvantifikaci „ne urologického“ selhání po užívá „Glasgow Coma Scale“
- c) kvantifikuje selhání celkem pěti orgánových systémů: kardi ovaskulárního, respiračního, hepatálního, renálního a hematologického
- d) kvantifikuje selhání celkem šesti orgánových systémů: kardi ovaskulárního, respiračního, hepatálního, renálního, hematologického a ne urologického (CNS)
4. V rámci kritického stavu může dojít k postižení nervového systému typu:
- a) encefalopati e
- b) polyne uropati e
- c) cévní mozkové příhody
- d) encefalitidy
- e) myelopati e
5. Encefalopati e kritického stavu:
- a) je označení pro encefalopatii provázející kritický stav s multi orgánovým selháním a SIRS, pokud jso u vylo učeny infekční encefalitida, parainfekční a uto imunitní encefalopati ea encefalopati e v důsledku primárního selhání některého z orgánů (játra, ledviny, srdce, plíce)
- b) je di agnostikována na základě EEG nálezu
- c) je di agnostikována na základě nálezu zobrazovacích metod
- d) manifestuje se typicky asymetrickými ložiskovými cerebrálními příznaky
- e) manifestuje se typicky poruchami kognitivních funkcí (pozornosti, paměti či dezori entací), rozvinutým deliri em přecházejícím v kvantitativní poruchu vědomí až do úrovně kómatu
6. Septická encefalopati e a infekční encefalitida se liší:
- a) přítomností pleocytózy u encefalitidy a její absencí u SE
- b) obvyklo u přítomností meninge álního syndromu u části nemocných s encefalitido u a jeho absencí u SE
- c) fokálními mozkovými příznaky u encefalitidy a jejích absencí u SE
- d) možno u přítomností epileptických záchvatů u encefalitidy a jejich absencí u SE
- e) charakteristickými abnormalitami zobrazovacích nálezů u SE
7. Parainfekční a uto imunitní encefalopati e (ADEM a AHLE) se od SE liší:
- a) specificko u EEG abnormalito u
- b) difuzními či víceložiskovými změnami bílé hmoty mozku v MR obraze
- c) histopatologickým obrazem perivenózního zánětu (ADEM) nebo vaskulitidy s fibrino idní nekrózo u (AHLE)
- d) přítomností frekventních epileptických záchvatů (postihujících > 25 % případů)
- e) fokálními mozkovými příznaky
8. Nově vzniklá svalová slabost u nemocných na JIP:
- a) je obvykle nervosvalového původu
- b) může být způsobena rozvojem Guillainova - Barrého syndromu, exacerbací preexistujícího nervosvalového onemocnění nebo perzistující blokádo u nervosvalového přenosu
- c) je nejčastěji způsobena ne uromuskulárními poruchami vznikajícími v so uvislosti s SIRS a multi orgánovým selháním: polyne uropati í kritického stavu, myopati í kritického stavu či jejich kombinací
- d) je vzácná; postihuje méně než 10 % kriticky nemocných
- e) je častá; postihuje více než 20 % kriticky nemocných
9. Perzistující nervosvalová blokáda:
- a) je přítomna po aplikaci blokátorů nervosvalového přenosu během po uze posledních 24 hod
b) manifestuje se generalizovano u svalovo u slabostí
c) manifestuje se areflexi í
d) je elektrofyzi ologicky detekovatelná pomocí dekrementu při repetitivní stimulaci motorického nervu
e) je doprovázena zvýšením kre atinkinázy v séru
10. Polyne uropati e kritického stavu:
- a) je výlučně axonální, převážně motorická či motoricko‑senzitivní
- b) může se manifestovat nově vzniklo u svalovo u slabostí
- c) může se manifestovat ztíženým odpojováním od ventilátoru
- d) u většiny případů jde o demyelinizační typ ne uropati e
- e) u většiny případů jde o senzitivní a/ nebo a utonomní typ ne uropati e
11. Elektrofyzi ologickým projevem polyne uropati e kritického stavu je:
- a) abnormální spontánní aktivita typu fibrilačních potenci álů a pozitivních ostrých vln
- b) snížením amplitud sumačního svalového akčního potenci álu (CMAP)
- c) snížením amplitud senzitivního nervového akčního potenci álu (SNAP)
- d) zpomalení rychlosti vedení motorickým nervem o více než 20 % pod normální limit
- e) nálezem snížené excitability sarkolemy při tzv. přímé svalové stimulaci
12. Myopati e kritického stavu:
- a) se projevuje nově vzniklo u svalovo u slabostí
- b) je dobře odlišitelná klinicky od polyne uropati e kritického stavu
- c) je zcela spolehlivě di agnostikovatelná po uze pomocí svalové bi opsi e
- d) je doprovázena zvýšením sérové hladiny kre atinkinázy (zejména nekrotická myopati e), avšak tento ukazatel není ani zcela senzitivní ani specifický pro CIM
- e) rutinní elektrofyzi ologické vyšetření nemusí přinést spolehlivo u diferenci aci oproti CIP, přínosná může být kvantitativní EMG (u ko operujícího paci enta) nebo přímá svalová stimulace
13. Atrofi e vláken typu 2:
- a) je jedním z možných histopatologických nálezů u myopati e kritického stavu
- b) je histopatologickým nálezem specifickým pro myopatii kritického stavu
- c) vyskytuje se kromě myopati e kritického stavu i u ne uropati í a stavů spojených s inaktivito u
- d) může být v rámci kritického stavu doprovázena ztráto u myosinových filament
- e) vylučuje di agnózu myopati e kritického stavu
14. Ztráta myosinových filament (Myosin Loss Myopathy):
- a) patří mezi charakteristické histopatologické nálezy u myopati e kritického stavu
- b) je histopatologická změna zcela charakteristická pro myopatii kritického stavu
- c) je prokazatelná re akcí s myosinovo u ATP - ázo u, imunohistochemicky pomocí protilátek proti myosinu či pomocí elektronmikroskopického vyšetření
- d) prokazuje se po uze pomocí elektronmikroskopického vyšetření
- e) vyskytuje se u myopati e kritického stavu po uze v důsledku předchozí aplikace kortikostero idů
15. Zvýšení kre atinkinázy v séru:
- a) je přítomno u více než poloviny nemocných s CIM, zejména v případě nekrotické myopati e
- b) může být přítomno i u nemocných s CIP
- c) umožňuje diferencovat mezi CIP a CIM
- d) je obvykle přechodné s maximemv prvním týdnu trvání kritickéhostavu a do 14 dnů se obvykle normalizuje
16. Polyne uromyopati e kritického stavu (CIPM):
- a) je samostatný typ ne uromuskulárního postižení v rámci kritického stavu zásadně odlišný od CIP a CIM
- b) je popisný termín vyjadřující často ukombinaci postižení kosterních svalůa periferního nervstva u kriticky nemocných
- c) manifestuje se zejména nově vzniklo u svalovo u slabostí
- d) je pozdní komplikací kritického stavu
- e) představuje selhání dalšího orgánu/ systému v rámci multi-orgánového selhání u kriticky nemocných
17. Nejčasnější elektrofyzi ologicko u abnormalito u signalizující rozvoj CIPM je:
- a) pokles amplitudy CMAP
- b) pokles amplitudy SNAP
- c) abnormální spontánní aktivita charakteru fibrilačních potenci álů a pozitivních ostrých vln
- d) simplifikace interferenčního vzorce
- e) dekrement při repetitivní stimulaci motorického nervu
18. Svalová bi opsi e:
- a) je indikována u všech nemocných s podezřením na CIP, CIM nebo CIPM
- b) je invazivní metodo u, která je v rutinní klinické praxi indikována po uze v případě, že je třeba od CIM odlišit jiné myopati e (zejména zánětlivo u myopatii)
- c) je nejspolehlivější metodo u detekce myogenního postižení u kriticky nemocných, a proto je so učástí nejnovější klasifikace myopati e kritického stavu
- d) má význam po uze v případě dostupnosti elektronové mikroskopi e
19. Prevence a léčba ne uromuskulárního postižení v rámci kritického stavu:
- a) je obdobně jako léčba septické encefalopati e prozatím ne uspokojivá
- b) v so učasnosti se zaměřuje na časno u a účinno u léčbu sepse (SIRS)
- c) zahrnuje snahu o eliminaci potenci álních rizikových faktorů, např. minimalizaci dávek kortikostero idů a NBNP
- d) zahrnuje co nejčasnější mobilizaci a komplexní rehabilitaci po odeznění kritického stavu
- e) zahrnuje blokádu efektu pro-zánětlivých cytokinů, která vedla k přesvědčivému efektu v léčbě CIP a CIM
20. Prognóza kriticky nemocných se známkami ne uromuskulárního postižení:
- a) je obecně dobrá; pokud nemocný přežije kritický stav, je obvyklá úplná úprava hybného deficitu
- b) je obecně špatná; mortalita kriticky nemocných je kolem 50 %
- c) po přežití kritického stavu může být relativně příznivá zejména u CIM
- d) je nepříznivě ovlivněna vznikem trvalého ne urologického deficitu,na kterém se kromě CIP a CIM mohou podílet i kompresivní mononeuropatie
správná je jedna nebo více odpovědí
Za správné vyřešení testu získá řešitel 5 kreditů ČLK.
Test můžete vyplnit na:
www.csnn.e u
Zdroje
1. Mahidhara R, Billi ar TR. Apoptosis in sepsis. Crit Care Med 2000; 28 (Suppl 4): N105 – N113.
2. Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunni on RE et al. Septic shock in humans. Advances in the understanding of pathogenesis, cardi ovascular dysfuncti on, and therapy. Ann Intern Med 1990; 113(3): 227 – 242.
3. Bone RC, Sprung CL, Sibbald WJ. Definiti ons for sepsis and organ failure. Crit Care Med 1992; 20(6): 724 – 726.
4. Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med 1996; 24(7): 1125 – 1128.
5. Casey LC, Balk RA, Bone RC. Plasma cytokine and endotoxin levels correlate with survival in pati ents with the sepsis syndrome. Ann Intern Med 1993; 119(8): 771 – 778.
6. Marano MA, Fong Y, Moldawer LL, Wei H, Calvano SE, Tracey KJ et al. Serum cachectin/ tumor necrosis factor in critically ill pati ents with burns correlates with infecti on and mortality. Surg Gynecol Obstet 1990; 170(1): 32 – 38.
7. Kna us WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of dise ase classificati on system. Crit Care Med 1985; 13(10): 818 – 829.
8. Kna us WA, Wagner DP, Draper EA, Zimmerman JE, Bergner M, Bastos PG et al. The APACHE III prognostic system. Risk predicti on of hospital mortality for critically ill hospitalized adults. Chest 1991; 100(6): 1619 – 1636.
9. Vincent JL, Moreno R, Takala J, Willatts S,de Mendonca A, Bruining H et al. The SOFA (Sepsis‑related Organ Failure Assessment) score to describe organ dysfuncti on/ failure. On behalf of the Working Gro up on Sepsis - Related Problems of the Europe an Soci ety of Intensive Care Medicine. Intensive Care Med 1996; 22(7): 707 – 710.
10. Vincent JL, de Mendonca A, Cantraine F, Moreno R, Takala J, Suter PM et al. Use of the SOFA score to assess the incidence of organ dysfuncti on/ failure in intensive care units: results of a multicenter, prospective study. Working gro up on „sepsis‑related problems“ of the Europe an Soci ety of Intensive Care Medicine. Crit Care Med 1998; 26(11): 1793 – 1800.
11. Moreno R, Vincent JL, Matos R, de Mendonca A, Cantraine F, Thijs L et al. The use of maximum SOFA score to qu antify organ dysfuncti on/ failure in intensive care. Results of a prospective, multicentre study. Working Gro up on Sepsis related Problems of the ESICM. Intensive Care Med 1999; 25(7): 686 – 696.
12. Janssens U, Graf C, Graf J, Radke PW, Königs B, Koch KC et al. Evalu ati on of the SOFA score: a single‑center experi ence of a medical intensive care unit in 303 consecutive pati ents with predominantly cardi ovascular disorders. Sequenti al Organ Failure Assessment. Intensive Care Med 2000; 26(8): 1037 – 1045.
13. Bednarik J, Vondracek P, Dusek L, Moravcova E, Cundrle I. Risk factors for critical illness polyne uromyopathy. J Ne urol 2005; 252(3): 343 – 351.
14. Bolton CF, Yo ung GB, Zochodne DW. The ne urological complicati ons of sepsis. Ann Ne urol 1993; 33(1): 94 – 100.
15. Chadwick J, Mann WN. Epidemics, Bo ok III. The medical works of Hippocrates. Oxford: Blackwell Sci entific Publicati ons 1950.
16. Eggers V, Schilling A, Kox WJ, Spi es C. Septic encephalopathy. Di agnosis und therapy. Anaesthesist 2003; 52(4): 294 – 303.
17. Eidelman LA, Putterman D, Putterman C, Sprung CL. The spectrum of septic encephalopathy. Definiti ons, eti ologi es, and mortaliti es. JAMA 1996; 275(6): 470 – 473.
18. Yo ung GB, Bolton CF, Austin TW, Archibald YM, Gonder J, Wells GA. The encephalopathy associ ated with septicillness. Clin Invest Med 1990; 13(6): 297 – 304.
19. Davi es NW, Shari ef MK, Howard RS. Infecti on‑associ ated encephalopathi es: their investigati on, di agnosis, and tre atment. J Ne urol 2006; 253(7): 833 – 845.
20. Yo ung GB, Bolton CF, Archibald YM, Austin TW, Wells GA. The electroencephalogram in sepsis‑associ ated encephalopathy. J Clin Ne urophysi ol 1992; 9(1): 145 – 152.
21. Za uner C, Gendo A, Kramer L, Funk GC, Ba uer E, Schenk P et al. Impaired subcortical and cortical sensory evoked potenti al pathways in septic pati ents. Crit Care Med 2002; 30(5): 1136 – 1139.
22. Bednařík J. Deliri um: nová výzva pro ne urologii? Cesk Slov Ne urol N 2006; 69/ 102(1): 18 – 26.
23. Kostalova M, Bednarik J, Haluzova A, Badurova R, Vohanka S, Urbanek I. Deliri um in stroke pati ents: epidemi ology and risk factors. J Ne urol 2007; 254 (Suppl 3): 115.
24. Papadopo ulos MC, Lamb FJ, Moss RF, Davi es DC, Tighe D, Bennett ED. Faecal peritonitis ca uses oedema and ne uronal injury in pig cerebral cortex. Clin Sci (Lond) 1999; 96(5): 461 – 466.
25. Dantzer R. Cytokine‑induced sickness behavi or: mechanisms and implicati ons. Ann N Y Acad Sci 2001; 933 : 222 – 234.
26. Dafny N, Pri eto - Gomez B, Dong WQ, Reyes - Vazquez C. Interferon modulates ne uronal activity recorded from the hypothalamus, thalamus, hippocampus, amygdala and the somatosensory cortex. Brain Res 1996; 734(1 – 2): 269 – 274.
27. Huynh HK, Dorovini‑Zis K. Effects of interferon - gamma on primary cultures of human brain microvessel endotheli al cells. Am J Pathol 1993; 142(4): 1265 – 1278.
28. Papadopo ulos MC, Davi es DC, Moss RF, Tighe D, Bennett ED. Pathophysi ology of septic encephalopathy: a revi ew. Crit Care Med 2000; 28(8): 3019 – 3024.
29. Ino uye SK, Viscoli CM, Horwitz RI, Hurst LD, Tinetti ME. A predictive model for deliri um in hospitalized elderly medical pati ents based on admissi on characteristics. Ann Intern Med 1993; 119(6): 474 – 481.
30. Marcantoni o ER, Goldman L, Mangi one CM, Ludwig LE, Muraca B, Hasla uer CM et al. A clinical predicti on rule for deliri um after elective noncardi ac surgery. JAMA 1994; 271(2): 134 – 139.
31. Eli e M, Cole MG, Prime a u FJ, Bellavance F. Deliri um risk factors in elderly hospitalized pati ents. J Gen Intern Med 1998; 13(3): 204 – 212.
32. Ino uye SK. Deliri um in older persons. N Engl J Med 2006 : 354(11): 1157 – 1165.
33. Marcantoni o ER, Rudolph JL, Culley D,Crosby G, Alsop D, Ino uye SK. Serum bi omarkers for deliri um. J Gerontol A Bi ol Sci Med Sci 2006; 61(12): 1281 – 1286.
34. Han L, McCusker J, Cole M, Abrahamowicz M, Prime a u F, Eli e M. Use of medicati ons with anticholinergic effect predicts clinical severity of deliri um symptoms in older medical inpati ents. Arch Intern Med 2001; 161(8): 1099 – 1105.
35. Flacker JM, Lipsitz LA. Serum anticholinergic activity changes with acute illness in elderly medical pati ents. J Gerontol A Bi ol Sci Med Sci 1999; 54(1): M12 – M16.
36. Flacker JM, Lipsitz LA. Ne ural mechanisms of deliri um: current hypotheses and evolving concepts. J Gerontol A Bi ol Sci Med Sci 1999; 54(6): B239 – B246.
37. Visvanathan K, Sundararajan V, Pugach P, Zabriski e JB. Postoperative cognitive decline: associ ati on with preoperative tumor necrosis factor‑alpha levels. J Am Geri atr Soc 2003; 51(11): 1673 – 1674.
38. Anzueto A. Muscle dysfuncti on in the intensive care unit. Clin Chest Med 1999; 20(2): 435 – 452.
39. Hopkins IJ. A new syndrome: poli omyelitis‑like illness associ ated with acute asthma in childho od. Aust Paedi atr J 1974; 10(5): 273 – 276.
40. Bednařík J, Vondráček P. Ne uromuskulární komplikace kritického stavu. Ne urol pro praxi 2001; 2 : 67 – 72.
41. Ambler Z. Diferenci ální di agnóza svalové slabosti u kriticky nemocných. Ne urol pro praxi 2001; 2 : 63 – 66.
42. Bizzarri - Schmid ND, Desai SP. Prolonged ne uromuscular blockade with atracuri um. Can Anaesth Soc J 1986; 33 : 209 – 212.
43. Pascuzzi RM. Evalu ati on and tre atment of pati ents with ne uromuscular juncti on disorders. In: American Academy of Ne urology (ed). 2000 Syllabi - on - CD - ROM. Northfi eld: Marathon Multimedi a 2000.
44. Adamus M, Bělohlávek R. Automatické dávkování svalových relaxanci í u náročných ne urochirurgických výkonů. Cesk Slov Ne urol N 2006; 69/ 102(6): 447 – 451.
45. Bednarik J. Critical illness polyne uropathy, myopathy or polyne uromyopathy: future directi ons. Int J Intens Care 2005; 12(1): 38 – 46.
46. Mertens HG. Di e dissemini erte ne uropathi e nach koma. Nervenarzt 1961; 32 : 71 – 79.
47. Henderson B, Koepke GH, Feller I. Peripheral polyne uropathy among pati ents with burns. Arch Phys Med Rehabil 1971; 52(4): 149 – 151.
48. Bolton CF, Brown JD, Sibbald WJ. The electrophysi ological investigati on of respiratory paralysis in critically ill pati ents. Ne urology 1983; 33 (Suppl 2): 186.
49. Roelofs RI, Cerra F, Bi elka N. Prolonged respiratory insuffici ency due to acute motor ne uropathy: a new syndrome. Ne urology 1983; 33 (Suppl 2): 240.
50. Bolton CF, Gilbert JJ, Hahn AF, Sibbald WJ. Polyne uropathy in critically ill pati ents. J Ne urol Ne urosurg Psychi atry 1984; 47(11): 1223 – 1231.
51. Hund EF. Ne uromuscular complicati ons in the ICU: the spectrum of critical illness‑related conditi ons ca using muscular we akness and we aning failure. J Ne urol Sci 1996; 136(1 – 2): 10 – 16.
52. Lopez Messa JB, García A. Acute polyne uropathy in critically ill pati ents. Intensive Care Med 1990; 16(3): 159 – 162.
53. Witt NJ, Zochodne DW, Bolton CF, Grand‘Maison F, Wells G, Yo ung GB et al. Peripheral nerve functi on in sepsis and multiple organ failure. Chest 1991; 99(1): 176 – 184.
54. Zochodne DW, Bolton CF, Wells GA, Gilbert JJ, Hahn AF, Brown JD et al. Critical illness polyne uropathy: a complicati on of sepsis and multiple organ failure. Brain 1987; 110 (Pt 4): 819 – 841.
55. Vondráček P, Bednařík J. Polyne uromyopati e kriticky nemocných – pilotní studi e. Cesk Slov Ne urol N 2000; 69/ 96(4): 226 – 233.
56. Tran DD, Groeneveld AB, van der Me ulen J, Na uta JJ, Strack van Schijndel RJ, Thijs LG. Age, chronic dise ase, sepsis, organ system failure, and mortality in a medical intensive care unit. Crit Care Med 1990; 18(5): 474 – 479.
57. Petersen B, Schneider C, Strassburg HM, Schrod L. Critical illness ne uropathy in pedi atric intensive care pati ents. Pedi atr Ne urol 1999; 21(4): 749 – 753.
58. Vondracek P, Bednarik J. Clinical and electrophysi ological findings and long‑term o utcomes in paedi atric pati ents with critical illness polyne uromyopathy. Eur J Paedi atr Ne urol 2006, 10(4): 176 – 181.
59. De Jonghe B, Sharshar T, Lefa uche ur JP, Authi er FJ, Durand - Zaleski I, Bo ussarsar M et al. Paresis acquired in the intensive care unit: a prospective multicenter study. JAMA 2002; 288(22): 2859 – 2867.
60. Wilmshurst PT, Tre acher DF, Lantos PL, Wiles CM. Critical illness polyne uropathy following severe hyperpyrexi a. QJM 1995; 88(5): 351 – 355.
61. Gla user MP, Zanetti G, Ba umgartner JD, Cohen J. Septic shock: pathogenesis. Lancet 1991; 338(8769): 732 – 736.
62. Low PA, Tuck RR, Take uchi M. Nerve microenvironment in di abetic polyne uropathy. In: Dyck PJ, Thomas PK, Asbury AK et al (eds). Di abetic Ne uropathy. Philadelphi a: WB Sa unders 1987 : 268 – 277.
63. Hund EF, Fogel W, Kri eger D, DeGeorgi a M, Hacke W. Critical illness polyne uropathy: clinical findings and o utcomes of a frequent ca use of ne uromuscular we aning failure. Crit Care Med 1996; 24(8): 1282 – 1283.
64. Gorson KC, Ropper AH. Acute respiratory failure ne uropathy: a vari ant of critical illness polyne uropathy. Crit Care Med 1993; 21(2): 267 – 271.
65. Wijdicks EF, Litchy WJ, Harrison BA, Gracey DR. The clinical spectrum of critical illness polyne uropathy. Mayo Clin Proc 1994; 69(10): 955 – 959.
66. Co akley JH, Nagendran K, Yarwo od GD, Honavar M, Hinds CJ. Patterns of ne urophysi ological abnormality in prolonged critical illness. Intensive Care Med 1998; 24(8): 801 – 817.
67. Schwarz J, Planck J, Bri egel J, Stra ube A. Single‑fiber electromyography, nerve conducti on studi es, and conventi onal electromyography in pati ents with critical - illness polyne uropathy: evidence for a lesi on of terminal motor axons. Muscle Nerve 1997; 20(6): 696 – 701.
68. Bednarik J, Vondracek P, Kadanka Z.Electrophysi ological differenti ati on between critical illness ne uropathy and myopathy: prospective case seri es study. Ne urology 2000; 54 (Suppl 3): A: 377.
69. Rich MM, Pinter MJ, Kraner SD, Barchi RL. Loss of electrical excitability in an animal model of acute qu adriplegic myopathy. Ann Ne urol 1998; 43(2): 171 – 179.
70. Rich MM, Teener JW, Raps EC, Schotland DL, Bird SJ. Muscle is electrically inexcitable in acute qu adriplegic myopathy. Ne urology 1996; 46(3): 731 – 736.
71. Rich MM, Bird SJ, Raps EC, McCluskey LF, Teener JW. Direct muscle stimulati on in acute qu adriplegic myopathy. Muscle Nerve 1997; 20(6): 665 – 673.
72. Bednařík J, Vondráček P, Lukáš Z, Dvořák K,Moravcová E, Adamová B et al. Polyne uromyopati e kritického stavu. I. Di agnostika. Cesk Slov Ne urol N 2002; 65/ 98(4): 386 – 394.
73. Bednarik J, Lukas Z, Vondracek P. Critical illness polyne uromyopathy: the electrophysi ological components of a complex entity. Intensive Care Med 2003; 29(9): 1505 – 1514.
74. Zifko UA. Long‑term o utcome of critical illness polyne uropathy. Muscle Nerve 2000; 23 (Suppl 9): S49 – S52.
75. Abraham E, Wunderink R, Silverman H,Perl TM, Nasraway S, Levy H et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in pati ents with sepsis syndrome. A randomized, controlled, do uble - blind, multicenter clinical tri al. TNF‑alpha MAb Sepsis Study Gro up. JAMA 1995; 273(12): 934 – 941.
76. Fisher CJ jr, Dhaina ut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ et al. Recombinant human interle ukin 1 receptor antagonist in the tre atment of pati ents with sepsis syndrome. Results from a randomized, do uble - blind, placebo - controlled tri al. Phase III rhIL‑1ra Sepsis Syndrome Study Gro up. JAMA 1994; 271(23): 1836 – 1843.
77. Zi egler EJ, Fisher CJ Jr, Sprung CL, Stra ube RC, Sadoff JC, Fo ulke GE et al.Tre atment of gram - negative bacteremi a and septic shock with HA - 1A human monoclonal antibody against endotoxin. A randomized, do uble - blind, placebo - controlled tri al. The HA - 1A Sepsis Study Gro up. N Engl J Med 1991; 324(7): 429 – 436.
78. Döcke WD, Randow F, Syrbe U, Kra usch D,Asadullah K, Reinke P et al. Monocyte de activati on in septic pati ents: restorati on by IFN - gamma tre atment. Nat Med 1997; 3(6): 678 – 681.
79. La upland KB, Kirkpatrick AW, Delaney A.Polyclonal intraveno us immunoglobulin for the tre atment of severe sepsis and septic shock in critically ill adults: A systematic revi ew and meta‑analysis. Crit Care Med 2007 Oct 23. [Epub ahe ad of print].
80. MacFarlane IA, Rosenthal FD. Severe myopathy after status asthmaticus. Lancet 1977; 2(8038): 615 – 620.
81. De Smet Y, Jaminet M, Jaeger U, Jacob J,Ne uray H, Ha us G et al. Acute corticostero id myopathy following status asthmaticus. Rev Ne urol (Paris) 1991; 147(10): 682 – 685.
82. Do uglass JA, Tuxen DV, Horne M, Scheinkestel CD, Weinmann M, Czarny D et al. Myopathy in severe asthma. Am Rev Respir Dis 1992; 146(2): 517 – 519.
83. Hirano M, Ott BR, Raps EC, Minetti C, Lennihan L, Libbey NP et al. Acute qu adriplegic myopathy: a complicati on of tre atment with stero ids, nondepolarizing blocking agents, or both. Ne urology 1992; 42(11): 2082 – 2087.
84. Lacomis D, Smith TW, Chad DA. Acute myopathy and ne uropathy in status asthmaticus: case report and literature revi ew. Muscle Nerve 1993; 16(1): 84 – 90.
85. Shapiro JM, Condos R, Cole RP. Myopathy in status asthmaticus: relati on to ne uromuscular blockade and corticostero id administrati on. J Intensive Care Med 1993; 8 : 144 – 152.
86. Barohn RJ, Jackson CE, Rogers SJ, Ridings LW, McVey AL. Prolonged paralysis due to non‑depolarizing ne uromuscular blocking agents and corticostero ids. Muscle Nerve 1994; 17(6): 647 – 654.
87. Van Marle W, Wo ods KL. Acute hydrocortisone myopathy. Br Med J 1980; 281(6235): 271 – 272.
88. Helliwell TR, Co akley JH, Wagenmakers AJ, Griffiths RD, Campbell IT, Green CJ et al. Necrotizing myopathy of critically - ill pati ents. J Pathol 1991; 164(4): 307 – 314.
89. Ramsay DA, Zochodne DW, Robertson DM, Nag S, Ludwin SK. A syndrome of acute severe muscle necrosis in intensive care unit pati ents. J Ne uropathol Exp Ne urol 1993; 52(4): 387 – 398.
90. Zochodne DW, Ramsay DA, Saly V, Shelley S, Moffatt S. Acute necrotizing myopathy of intensive care: electrophysi ological studi es. Muscle Nerve 1994; 17(3): 285 – 292.
91. al - Lozi MT, Pestronk A, Yee WC, Flairs N,Co oper J. Rapidly evolving myopathy with myosin defici ent muscle fibers. Ann Ne urol 1994; 35(3): 273 – 279.
92. Sher JH, Shafiq SA, Schutta HS. Acute myopathy with selective lysis of myosin filaments. Ne urology 1979; 29(1): 100 – 106.
93. Gorson KC, Ropper AH. Generalized paralysis in the intensive care unit: emphasis on the complicati ons of ne uromuscular blocking agents and corticostero ids. J Int Care Med 1996; 11 : 219 – 231.
94. Hund EF. Myopathy in critically ill pati ents. Crit Care Med 1999; 27 : 2544 – 2547.
95. Deconinck N, Van Parijs V, Beckers--Ble ukx G, Van den Bergh P. Critical illness myopathy unrelated to corticostero ids or ne uromuscular blocking agents. Ne uromusc Disord 1998; 8 : 186 – 192.
96. Helliwell TR, Wilkinson A, Griffiths RD, McClelland P, Palmer TE, Bone JM. Muscle fibre atrophy in critically ill pati ents is associ ated with the loss of myosin filaments and the presence of lysosomal enzymes and ubiquitin. Ne uropathol Appl Ne urobi ol 1998; 24 : 507 – 517.
97. De Letter MA, van Do orn PA, Savelko ul HF, Laman JD, Schmitz PI, Op de Co ul AA et al. Critical illness polyne uropathy and myopathy (CIPNM): evidence for local immune activati on by cytokine - expressi on in the muscle tissue. J Ne uro immunol 2000; 106 : 206 – 213.
98. Lacomis D, Petrella JT, Gi uli ani MJ. Ca uses of ne uromuscular we akness inthe intensive care unit: a study of ninety - two pati ents. Muscle Nerve 1998; 21 : 610 – 617.
99. Latronico N, Fenzi F, Recupero D, Gu arneri B, Tomelleri G, Tonin P et al. Critical illness myopathy and ne uropathy. Lancet 1996; 347 : 1579 – 1582.
100. Ruff RL. Acute illness myopathy. Ne urology 1996; 46 : 600 – 601.
101. DuBo is DC, Almon RR. A possible role for glucocortico ids in denervati on atrophy. Muscle Nerve 1981; 4 : 370 – 373.
102. Ro ule a u G, Karpati G, Carpenter S, Soza M, Prescott S, Holland P. Glucocortico id excess induces preferenti al depleti on of myosin in denervated skeletal muscle fibers. Muscle Nerve 1987; 10 : 428 – 438.
103. Wing SS, Goldberg AL. Glucocortico ids activate the ATP - ubiquitin‑dependent proteolytic system in skeletal muscle during fasting. Am J Physi ol 1993; 264 : 668 – 676.
104. Showalter CJ, Engel AG. Acute qu adriplegic myopathy: analysis of myosin isoforms and evidence of calpain‑medi ated proteolysis. Muscle Nerve 1997; 20 : 316 – 322.
105. Raps EC, Bird SJ, Hansen - Flaschen JH. Prolonged muscle we akness after ne uromuscular blockade in the intensive care unit. Crit Care Clinics 1994; 10 : 799 – 813.
106. Waclawik AJ, Sufit RL, Beinlich BR, Schutta HS. Acute myopathy with selective degenerati on of myosin filaments following status asthmaticus tre ated with methylprednisolone and vecuroni um. Ne uromusc Disord 1992; 2 : 19 – 26.
107. Kupfer Y, Okrent DG, Twersky RA, Tessler S. Disuse atrophy in a ventilated pati ent with status asthmaticus receiving ne uromuscular blockade. Crit Care Med 1987; 15 : 795 – 796.
108. Meyer KC, Pri elipp RC, Grossman JE, Co ursin DB. Prolonged we akness after infusi on of atracuri um in two intensive care unit pati ents. Anesth Analg 1994; 78 : 772 – 774.
109. Bolton CF. Sepsis and the systemic inflammatory response syndrome: Ne uromuscular manifestati ons. Crit Care Med 1996; 24 : 1408 – 1416.
110. Clowes GHA, George BC, Villee CA. Muscle proteolysis induced by a circulating peptide in pati ents with sepsis or tra uma. N Engl J Med 1983; 308 : 545 – 552.
111. Apte - Kakade F. Rehabilitati on of pati ents with qu adriparesis after tre atment of status asthmaticus with ne uromuscular blocking agents and high dose corticostero ids. Arch Phys Med Rehabil 1991; 72 : 1024 – 1028.
112. Sitwell LD, Weinshenker BG, Monpetit V, Reid D. Complete ophthalmoplegi a as a complicati on of acute corticostero id and pancuroni um associ ated myopathy. Ne urology 1991; 41 : 921 – 922.
113. Campellone JV, Lacomis D, Kramer DJ, van Cott AC, Gi uli ani MJ. Acute myopathy after liver transplantati on. Ne urology 1998; 50 : 46 – 53.
114. Lacomis D, Gi uli ani MJ, Van Cott A, Kramer DJ. Acute myopathy of intensive care: clinical, electromyographic, and pathological aspects. Ann Ne urol 1996; 40 : 645 – 654.
115. Yarom R, Sphira Y. Myosin degenerati on in a congenital myopathy. Arch Ne urol 1977; 34 : 114 – 115.
116. Bazzi P, Moggi o M, Prelle A, Sci acco M,Messina S, Barbi eri S et al. Critically ill pati ents: immunological evidence of inflammati on in muscle bi opsy. Clin Ne uropathol 1999; 18 : 23 – 30.
117. Lacomis D, Zochodne DW, Bird SJ. Critical illness myopathy. Muscle Nerve 2000; 23 : 1785 – 1788.
118. Co akley J. Sho uld ICU pati ents receive muscle relaxants? Schweiz Med Wochenschr 1996; 126 : 1644 – 1648.
119. Op de Co ul AAW, Lambregts PCLA, Koeman J, van Puyenbroek PJ, Ter Laak HJ, Gabreels - Festen AA. Ne uromuscular complicati ons in pati ents give Pavulon (pancuroni um bromide) during artifici al ventilati on. Clin Ne urol Ne urosurg 1985; 87 : 17 – 22.
120. Op de Co ul AA, Verhe ul GA, Leyten AC, Schellens RL, Teepen JL. Critical illness polyne uromyopathy after artifici al respirati on. Clin Ne urol Ne urosurg 1991; 93 : 27 – 33.
121. Latronico N, Bertolini G, Gu arneri B, Botteri M, Peli E, Andreoletti S et al. Simplifi ed electrophysi ological evalu ati on of peripheral nerves in critically ill pati ents: the Itali an multi - centre CRIMYNE study. Crit Care 2007; 11: R11.
122. Trojaborg W, Weimer LH, Hays AP. Electrophysi ologic studi es in critical illness associ ated we akness: myopathy or ne uropathy – a re appraisal. Clin Ne urophysi ol 2001; 112 : 1568 – 1593.
123. Latronico N. Ne uromuscular alterati ons in the critically ill pati ent: critical illness myopathy, critical illness ne uropathy, or both? Intensive Care Med 2003; 29 : 1411 – 1413.
124. De Letter MA, Schmitz PI, Visser LH, Verhe ul FA, Schellens RL, Op de Co ul DA et al. Risk factors for the development of polyne uropathy and myopathy in critically ill pati ents. Crit Care Med 2001; 29 : 2281 – 2286.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2008 Číslo 5
Nejčtenější v tomto čísle
- Neurologické poruchy v rámci kritického stavu
- Intradurální výhřez bederní meziobratlové ploténky manifestující se syndromem kaudy – kazuistika
- Současná diagnostika a léčba oligodendrogliomů
- Traumatické poranění mozku a zlomeniny obličejového skeletu