
Novel familial SGCE gene variant associated with myoclonus-dystonia and concomitant multiple sclerosis?
Authors:
M. Wayhelová 1; I. Šrotová 2,3; E. Minks 4,5; J. Martinková 1; A. Křepelová 1; M. Macek Jr 1; S. Flašarová 3; M. Hladíková 2,3; M. Petrášová 2,3; E. Vlčková 2,3; P. Štourač 2,3
Authors place of work:
Department of Biology and Medical Genetics, Second Faculty of Medicine, Charles University and Motol University Hospital, Prague, Czech Republic
1; Faculty of Medicine, Masaryk University Brno, Brno, Czech Republic
2; Department of Neurology, Faculty of Medicine, Masaryk University, University Hospital Brno, Brno, Czech Republic
3; First Department of Neurology, St. Anne‘s University Hospital Brno, Brno, Czech Republic
4; Neurology, TERAneuro s. r. o., Židlochovice, Czech Republic
5
Published in the journal:
Cesk Slov Neurol N 2026; 89(1): 50-52
Category:
Dopis redakci
doi:
https://doi.org/10.48095/cccsnn202650
Myoclonus-dystonia (MD) is a movement disorder characterized by a combination of rapid, brief muscle contractions (myoclonus). The myoclonic jerks typically affect the neck, trunk, and upper limbs, with less common involvement of the legs. Approximately 50% of the affected individuals have additional focal or segmental dystonia, presenting as cervical dystonia and/or writer's cramps. Non-motor features may include an increased risk of alcohol abuse, obsessive-compulsive disorder, and anxiety disorders [1]. Symptom onset is usually in the first decade of life and almost always by the age of 20 years. MD is an extremely rare disease, with an estimated prevalence of one to two cases per million people [2]. Pathogenic variants in ξ-sarcoglycan gene SGCE represent the most frequent genetic cause of MD with maternal imprinting. Therefore, the clinical manifestation is dependent on the transmission of the pathogenic variants in the paternal lineage [3]. The clinical diagnosis of MD is then confirmed by the presence of a heterozygous pathogenic variant in the SGCE gene.
All procedures including clinical and molecular diagnostics and therapy were conducted as a part of routine diagnostics in accordance with the principles of the Declaration of Helsinki. The written institutional informed consent was obtained from all participants before the genetic analysis. The manuscript does not contain any photographs, videos or information of any recognizable persons.
A male proband had a history of myoclonic jerks of the left upper limb and leg, focal cervical dystonia, and writer´s cramp. First symptoms appeared at the age of 12 and did not progress over time and did not interfere with most of his daily activities. Similar symptoms were already present (or later developed) in his father, sister and one of his daughters (Tab. 1). At the age of 33, the patient developed also panic attacks and anxiety disorder, probably also related to MD. Therefore, the highly specific phenotype was then indicative for the molecular genetic analysis of the SGCE gene of which rare pathogenic variants are well recognized as the primary molecular cause of MD [4].
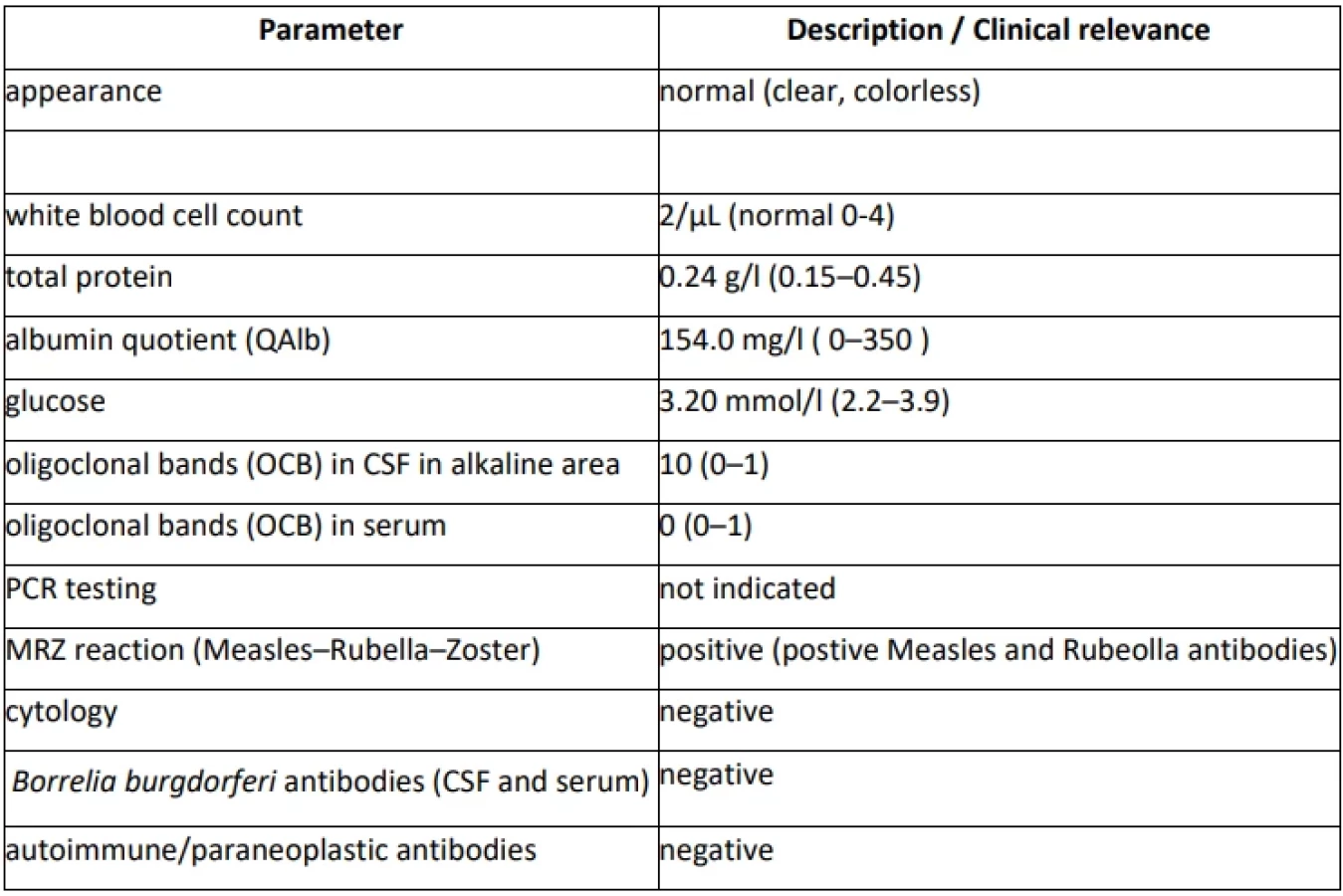
At the age of 48, new symptoms of weakness and paresthesia in the left-sided extremities newly developed within a week. Magnetic resonance (MRI) showed the presence of supratentorial (T2-hyperintense periventricular and 2 juxtacortical lesions) and infratentorial brain lesions and one spinal cord lesion at C3/C4 level susceptive of multiple sclerosis (MS). A cerebrospinal fluid (CSF) examination revealed the presence of 10 oligoclonal bands (OCBs), while no OCBs were found in corresponding serum (Supplementary Table S1). The diagnosis of MS was established, and the patient was treated with high-dose steroids and ofatumumab (Tab. 1). The clinical symptoms of MS completely resolved within two weeks. Follow-up brain MRI showed no new lesions or progression of the already described ones, moreover, it showed a reduction of one lesion present in the diagnostic brain MRI. Patient has adhered to the therapy and tolerated it well (see diagnostic and therapeutic workflow in Fig. 1).


The molecular genetic analyses of the SGCE gene were performed in the proband and his affected relatives (Figure 2). Genomic DNA was extracted using the MagCore® Genomic DNA Large Volume Whole Blood Kit on MagCore® Super/HF 16 Plus Automated Nucleic Acid Extractor (RBC Bioscience Corporation, New Taipei City, Taiwan). The following molecular genetic analysis by Sanger sequencing was initially indicated in proband’s daughter and the segregation of the candidate causative variant was then tested in the proband, his sister and father.

Molecular genetic analysis by Sanger sequencing including PCR with custom primers (Supplementary Table S2) followed by termination reaction using BigDyeTM Terminator v3.1 Cycle Sequencing Kit was performed according to the manufacturer’s recommendation (Applied Biosystems, Waltham, MA, USA). The samples were run on ABI 3130xl and SeqStudio Flex Genetic Analyzers (Applied Biosystems). Chromatograms were analyzed using Chromas Lite (Technelysium Pty Ltd, South Brisbane, Australia).
Afterwards, RNA from the peripheral blood of the proband and healthy unrelated individual (control sample) was extracted using the PAXgene Blood RNA System (PreAnalytiX, Hombrechtikon, Switzerland). The one-step cDNA synthesis and PCR amplification with custom primers was performed using the SuperScriptTM III One-Step RT-PCR System with PlatinumTM Taq DNA-Polymerase (Applied Biosystems). The integrity of both RNA samples was checked using the endogenous control primer pair for the GCH1 mRNA and RT-PCR products were detected in the proband‘s and control samples. The primer pairs were designed to amplify the region of interest of the SGCE gene including the borderline between the exon 5 and 6 and the control region encompassing the exon 2 to 4 (Supplementary Table S2).

The pathogenicity of the candidate variant was evaluated according to the recommendations of the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) [5].
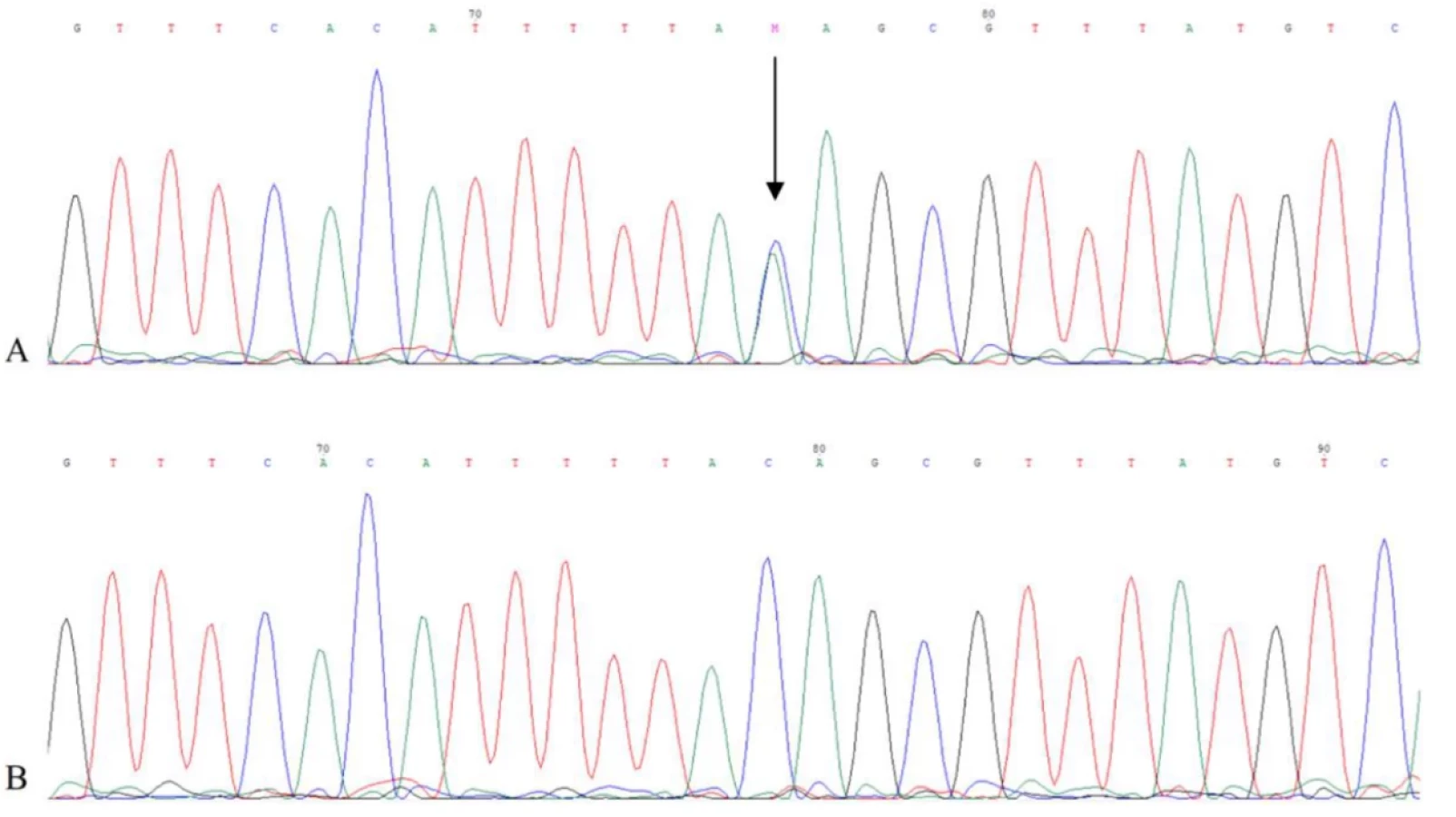
The genetic analysis of the SGCE gene identified a heterozygous variant NC_000007.14(NM_003919.2):c.663-3C>A in the intron 5 of the SGCE gene and, hence, confirmed the diagnosis of MD when the patient was 47 years old (Figure 3). Its deleterious effect on splicing was predicted using in silico tools SpliceAI (output: D type: acceptor loss, D score: 0.87, position: -3 bp) (https://spliceailookup.broadinstitute.org) and Human Splicing Finder (HSF) (output: Broken WT Acceptor Site: Alteration of the WT Acceptor site, most probably affecting splicing) (https://genomnis.com/hsf). The subsequent RT-PCR confirmed the deleterious effect on splicing as the region of interest and the control region of the SGCE gene was amplified in an RNA sample from a healthy unrelated individual, but not from the patient. Finally, the variant was submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) under the Variation ID: 3912069 and to the Leiden Open Variation Database (https://www.lovd.nl) under the DB-ID: SGCE_000113.
We report on the case of a rare coincidence of MS and MD. The patient presented in the case report has had symptoms of MD since his childhood, with long-term stable course. However, the phenotypic manifestation of MD has been observed in his family in the paternal lineage, and therefore suggestive for molecular genetic analysis of the SGCE gene.
The SGCE gene (MIM:604149) encodes ξ-sarcoglycan, a member of the sarcoglycan family. Sarcoglycans are a part of the dystrophin-glycoprotein complex [6]. It is widely expressed in multiple parts of the central nervous system. The SGCE gene is a subject of a genomic imprinting with paternal-specific expression explaining the autosomal dominant inheritance with reduced penetrance [7]. To this date, 45 isoforms have been identified as the result of the SGCE pre-mRNA alternative splicing. The deficiency of the major isoform e-SG1 and other isoforms including the “brain-specific” exon 11b is confirmed as the major genetic contributor in the pathogenesis of MD [8]. Across the years, multiple rare and recurrent loss-of-function variants in the SGCE gene have been linked to MD [9,10].
Therefore, the novel splicing variant NM_003919.2:c.663-3C>A on the borderline of the intron 5 and exon 6 of the SGCE was confirmed as the molecular cause of MD in affected individuals in the reported family. To our best knowledge, this variant has not been identified and associated with MD so far. We concluded that the variant was likely responsible for the aberrant splicing. The definitive proof of its pathogenicity was given by the sequencing analysis of the control region and the region of interest on the proband`s RNA. The aberrant transcripts expressed from the mutated paternal allele are likely completely degraded by the nonsense-mediated mRNA decay, leading to the loss of expression of the paternal allele and the absence of the functional protein ξ-sarcoglycan. The experimental outputs are in full accordance with outputs of in silico prediction tools (SpliceAI and HSF). The aberrant splicing leading to the partial or complete degradation of the altered transcripts of the paternal SGCE allele is a relatively common mechanism, causing the absence of a functional ξ-sarcoglycan as there are more than 25 different causative splicing variants in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar; accessed 2025-09-12) and multiple cases published.
SGCE‑related MD comprises of 30–50 % of all clinically defined cases of MD with a typical onset in the childhood or adolescence (3-10 years of age) [11]. Due to the autosomal dominant inheritance with maternal imprinting and frequent milder or atypical clinical manifestation the affected individuals are underdiagnosed, therefore the real incidence might be higher than reported elsewhere [2]. According to the best authors´ knowledge, this is the first published report of the coincidence of MS and MD. While MD is an extremely rare disease, the global prevalence of MS reaches 23.9 cases per 100 000 population, with a continuous increase over the past three decades [12].
Genetic confirmation of MD did not come until the patient was 47 years old. A year later, new neurological problems emerged within a few days, which he addressed with his treating neurologist, and which were later attributed to MS. The main challenge in similar situations is to distinguish if the new symptoms represent a complication or progression of the already known neurological condition or if there is a new neurological disease with a chance of co-incidence with the previously known disease.
In our case report, the latter option was more probable since motor and sensory symptoms do not represent a common clinical manifestation of MD. For this reason, the brain and cervical spinal cord MRI was performed. Patients with MD usually have a normal brain MRI [7]. Specific white matter abnormalities in MD can occur, but mainly apparent in the functional MRI of the subthalamic region of the brainstem [13]. The brain and cervical spinal cord MRI showed several supra - and infratentorial and spinal cord demyelinating lesions which fulfilled 2017 McDonald's criteria for MS [14]. The CSF examination was performed with the finding of OCBs in CSF (not present in serum) supportive of the MS diagnosis. The normal CSF finding with low levels of the serotonin metabolite, 5-hydroxyindoleacetic acid, is typical for MD (not tested in proband) [15]. Based on radiological findings and negative clinical prognostic factors, highly effective corticosteroid therapy has been initiated. Motor and sensory symptoms of MS have completely resolved while MD remained unchanged. Patient´s psychiatric symptoms are probably part of MD, although they may also be present in patients with MS [1,16].
The random co-occurrence of two independent diagnostic units, i.e. MD and MS, in our patient was thus the most probable explanation of his clinical problems.
To conclude, the familial segregation of MD as the consequence of a novel deleterious splicing variant NM_003919.2(SGCE):c.663-3C>A extends the current spectrum of clinically relevant SGCE gene variants and point out the usefulness and necessity of complementary RNA analysis from the peripheral blood for the assessment of the pathogenicity of candidate SGCE gene variants. The presence of two independent clinical entities in the proband should be reflected for the initiation of the appropriate therapy, if applicable, and long-term medical follow-up.
Funding
This work was supported by the Specific research project no. MUNI/A/1186/2022 provided by Masaryk University Brno, and by the Ministry of Health of the Czech Republic project for conceptual development in research organizations ref. no. 65269705 (University Hospital Brno, Brno, Czech Republic), MH CZ-DRO, Motol University Hospital, Prague, Czech Republic 00064203 (6003) and NCMG (The National Center for Medical Genomics; ncmg.cz), LM2018132.
Conflict of Interest Statement
The authors declare no conflict of interest.
Zdroje
1. Hess CW, Raymond D, Aguiar Pde C et al. Myoclonus-dystonia, obsessive-compulsive disorder, and alcohol dependence in SGCE mutation carriers. Neurology 2007; 68(7): 522–524. doi: 10.1212/01.wnl.0000253188.76092.06.
2. Yilmaz Z, Peall K. THUR 104 Global MD registry and dystonia non-motor symptoms study. J Neurol Neurosurg Psychiatry 2018; 89 (suppl 1): A11.2. doi: 10.1136/jnnp-2018-abn.40.
3. Grabowski M, Zimprich A, Lorenz-Depiereux B et al. The epsilon-sarcoglycan gene (SGCE), mutated in myoclonus-dystonia syndrome, is maternally imprinted. Eur J Hum Genet 2003; 11(2): 138–144. doi: 10.1038/sj.ejhg.5200938.
4. Cazurro-Gutiérrez A, Marcé-Grau A, Correa-Vela M et al. ε-Sarcoglycan: unraveling the myoclonus-dystonia gene. Mol Neurobiol 2021; 58(8): 3938–3952. doi: 10.1007/s12035-021-02391-0.
5. Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17(5): 405–424. doi: 10.1038/gim.2015.30.
6. Esapa CT, Waite A, Locke M et al. SGCE missense mutations that cause myoclonus-dystonia syndrome impair epsilon-sarcoglycan trafficking to the plasma membrane: modulation by ubiquitination and torsinA. Hum Mol Genet 2007; 16(3): 327–342. doi: 10.1093/hmg/ddl472.
7. Müller B, Hedrich K, Kock N et al. Evidence that paternal expression of the epsilon-sarcoglycan gene accounts for reduced penetrance in myoclonus-dystonia. Am J Hum Genet 2002; 71(6): 1303–1311. doi: 10.1086/344531.
8. Xiao J, Vemula SR, Xue Y et al. Role of major and brain-specific Sgce isoforms in the pathogenesis of myoclonus-dystonia syndrome. Neurobiol Dis 2017; 98 : 52–65. doi: 10.1016/j.nbd.2016.11.003.
9. Zimprich A, Grabowski M, Asmus F et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet 2001; 29(1): 66–69. doi: 10.1038/ng709.
10. Peall KJ, Kurian MA, Wardle M et al. SGCE and myoclonus dystonia: motor characteristics, diagnostic criteria and clinical predictors of genotype. J Neurol 2014; 261(12): 2296–2304. doi: 10.1007/s00415-014-7488-3.
11. Mencacci N, R'bibo L, Bandres-Ciga S et al. The CACNA1B R1389H variant is not associated with myoclonus-dystonia in a large European multicentric cohort. Hum Mol Genet 2015; 24(18): 5326–5329. doi: 10.1093/hmg/ddv255.
12. Khan G, Hashim MJ. Epidemiology of multiple sclerosis: global, regional, national and sub-national-level estimates and future projections. J Epidemiol Glob Health 2025; 15(1): 21. doi: 10.1007/s44197-025-00353-6.
13. van der Meer JN, Beukers RJ, van der Salm SM et al. White matter abnormalities in gene-positive myoclonus-dystonia. Mov Disord 2012; 27(13): 1666–1672. doi: 10.1002/mds.25128.
14. Thompson AJ, Banwell BL, Barkhof F et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018; 17(2): 162–173. doi: 10.1016/S1474-4422(17)30470-2..
15. Peall KJ, Ng J, Dy ME et al. Low CSF 5-HIAA in myoclonus dystonia. Mov Disord 2017; 32(11): 1647–1649. doi: 10.1002/mds.27117.
16. Peall KJ, Waite AJ, Blake DJ et al. Psychiatric disorders, myoclonus dystonia, and the epsilon-sarcoglycan gene: a systematic review. Mov Disord 2011; 26(10):1939-42. doi: 10.1002/mds.23791.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2026 Číslo 1
Nejčtenější v tomto čísle
- Guidelines of the Czech Neurological Society of the CMA JEP and the Czech Neurosurgical Society of the CMA JEP for the management of spontaneous intracerebral hemorrhage in adult patients – version 2026
- Self-management in adult patients after stroke – a review of self-management programs
- Ladislav Haškovec and establishing the University Department for Nervous Diseases of Charles University
- Ofatumumab in the treatment of multiple sclerosis – from clinical outcomes to the economic sustainability of treatment